| Journal of Medical Cases, ISSN 1923-4155 print, 1923-4163 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Med Cases and Elmer Press Inc |
| Journal website https://jmc.elmerpub.com |
Case Report
Volume 16, Number 5, May 2025, pages 164-173

Nephronophthisis and Retinitis Pigmentosa (Senior-Loken Syndrome) After Living-Donor Kidney Transplantation: Twelve-Year Follow-Up in a Young Woman
Toshihiko Matsuoa, b, f, Yasuhiro Onishic, Hiroshi Morinagac, Jun Wadac, Takehiro Tanakad, Motoo Arakie
aGraduate School of Interdisciplinary Science and Engineering in Health Systems,
Okayama University, Okayama City 700-8558, Japan
bDepartment of Ophthalmology,
Okayama University Hospital, Okayama City 700-8558, Japan
cDepartment of
Nephrology, Rheumatology, Endocrinology and Metabolism, Graduate School of Medicine, Dentistry,
and Pharmaceutical Sciences, Okayama University, Okayama City 700-8558,
Japan
dDepartment of Pathology, Graduate School of Medicine, Dentistry, and
Pharmaceutical Sciences, Okayama University, Okayama City 700-8558,
Japan
eDepartment of Urology, Graduate School of Medicine, Dentistry, and
Pharmaceutical Sciences, Okayama University, Okayama City 700-8558,
Japan
fCorresponding Author: Toshihiko Matsuo, Regenerative and Reconstructive
Medicine (Ophthalmology), Graduate School of Interdisciplinary Science and Engineering in Health
Systems, Okayama University, Okayama City 700-8558, Japan
Manuscript submitted January 25, 2025, accepted May 12, 2025, published online May 28,
2025
Short title: Senior-Loken Syndrome After Kidney Transplant
doi:
https://doi.org/10.14740/jmc4356
| Abstract | ▴Top |
Senior-Loken syndrome is a hereditary ciliopathy with recessive trait that manifests as nephronophthisis and retinitis pigmentosa. This report described an 18-year-old woman who was referred to a University Hospital to set up a treatment plan for chronic renal failure of an unknown cause. She had experienced nocturnal polyurea from the age of 12 years and was found to have an elevated level of serum creatinine at 3 mg/dL at the age of 15 years. She underwent renal biopsy at a hometown regional hospital which showed global glomerulosclerosis in six of the 13 glomeruli examined, renal tubular dilation in irregular shape, and marked interstitial fibrosis with lymphocytic infiltration. At the age of 19 years, she received a living-donor kidney transplant from her 46-year-old father as a preemptive therapy. At surgery, biopsy of the father’s donor kidney showed two glomeruli with global sclerosis out of 24 glomeruli examined, in association with minimal interstitial fibrosis and lymphocytic infiltration. She began to have extended-release tacrolimus 4 mg daily and mycophenolate mofetil 1,000 mg daily. According to the standard protocol, she underwent biopsy of the transplanted donor kidney to reveal interstitial fibrosis and lymphocytic infiltration, in addition to no sign of rejection and no glomerular deposition of immunoglobulins and complements, both 4 weeks and 14 months after the kidney transplantation. At the age of 23 years, 4 years after the kidney transplantation, she was, for the first time, diagnosed retinitis pigmentosa, and hence, Senior-Loken syndrome. She was followed up in the stable condition with basal doses of tacrolimus 5 mg daily, mycophenolate mofetil 1,000 mg daily, and prednisolone 5 mg daily up until now in 12 years after the kidney transplantation. The interstitial fibrosis with lymphocytic infiltration in the donor kidney might be a milder presentation of the disease with recessive inheritance.
Keywords: Retinitis pigmentosa; Nephronophthisis; Senior-Loken syndrome; Kidney transplantation; Living donor; Kidney biopsy; Pathology; Computed tomography scan; Ciliopathy; Optical coherence tomography
| Introduction | ▴Top |
Retinitis pigmentosa is a hereditary disease that shows varying degrees of retinal dystrophy usually with a symptom of night blindness [1]. The visual field constriction, often in the form of concentric narrowing in both eyes, is caused by the different extent of retinal involvement with dystrophy, and the speed of deterioration in dystrophy varies largely from individual to individual. Retinitis pigmentosa which occurs in association with other systemic diseases is designated as syndromic retinitis pigmentosa [2]. Major underlying causes for syndromic retinitis pigmentosa belong to so-called ciliopathies which are attributed to abnormal function or structure of primary cilia of cells. Retinal photoreceptor cells consist of inner segments with cell bodies and outer segments with disc membranes, both of which are joined by connecting cilia as classified as primary cilia. Well-known entities of syndromic retinitis pigmentosa as ciliopathies are Usher syndrome which shows hearing disturbance and Bardet-Biedl syndrome which manifests hypogonadism and obesity [2].
Ciliopathies also occur in the kidney since renal tubular epithelial cells have primary cilia [3]. Two major types of ciliopathies in the kidney are polycystic kidney disease and nephronophthisis. Polycystic kidney disease shows a dominant or recessive trait [3, 4]. In contrast, nephronophthisis is a recessive disease with mutations in genes designated as series of NPHP or nephrocystins to cause end-stage renal disease in children, juveniles, and adolescents [5, 6]. Nephronophthisis that is associated with retinitis pigmentosa is called Senior-Loken syndrome [7, 8]. In this study, we presented a young woman who developed chronic renal failure and received a living-donor kidney transplant from her father as a preemptive therapy. She was diagnosed as retinitis pigmentosa 4 years later, and hence was designated as Senior-Loken syndrome.
| Case Report | ▴Top |
An 18-year-old woman was referred to a University Hospital to set up a treatment plan for chronic renal failure since she moved to this city from her hometown to enter a college. She had experienced nocturnal polyurea and had been taking oral iron preparation from the age of 12 years. At 15 years old, she developed low-grade fever after an overseas travel to attend a summer school and was found to have an elevated level of serum creatinine at 3 mg/dL at a hometown regional hospital. She underwent renal biopsy at 16 years old in that hospital: of the 13 glomeruli examined, approximately half (6/13) showed global sclerosis, and one-quarter (3/13) showed atubular glomeruli. Severe interstitial fibrosis and tubular atrophy with irregular dilation were observed, along with focal cellular infiltration (Fig. 1a, b). A definite pathological diagnosis was not reached at that time. She was referred to another regional hospital and began to have oral prednisolone tapering from 30 mg daily in a suspected diagnosis of chronic tubulointerstitial nephritis. In family history, her paternal female cousin died of renal failure at the age of 22 years. There was no consanguineous marriage in the family. Her two younger sisters were healthy.
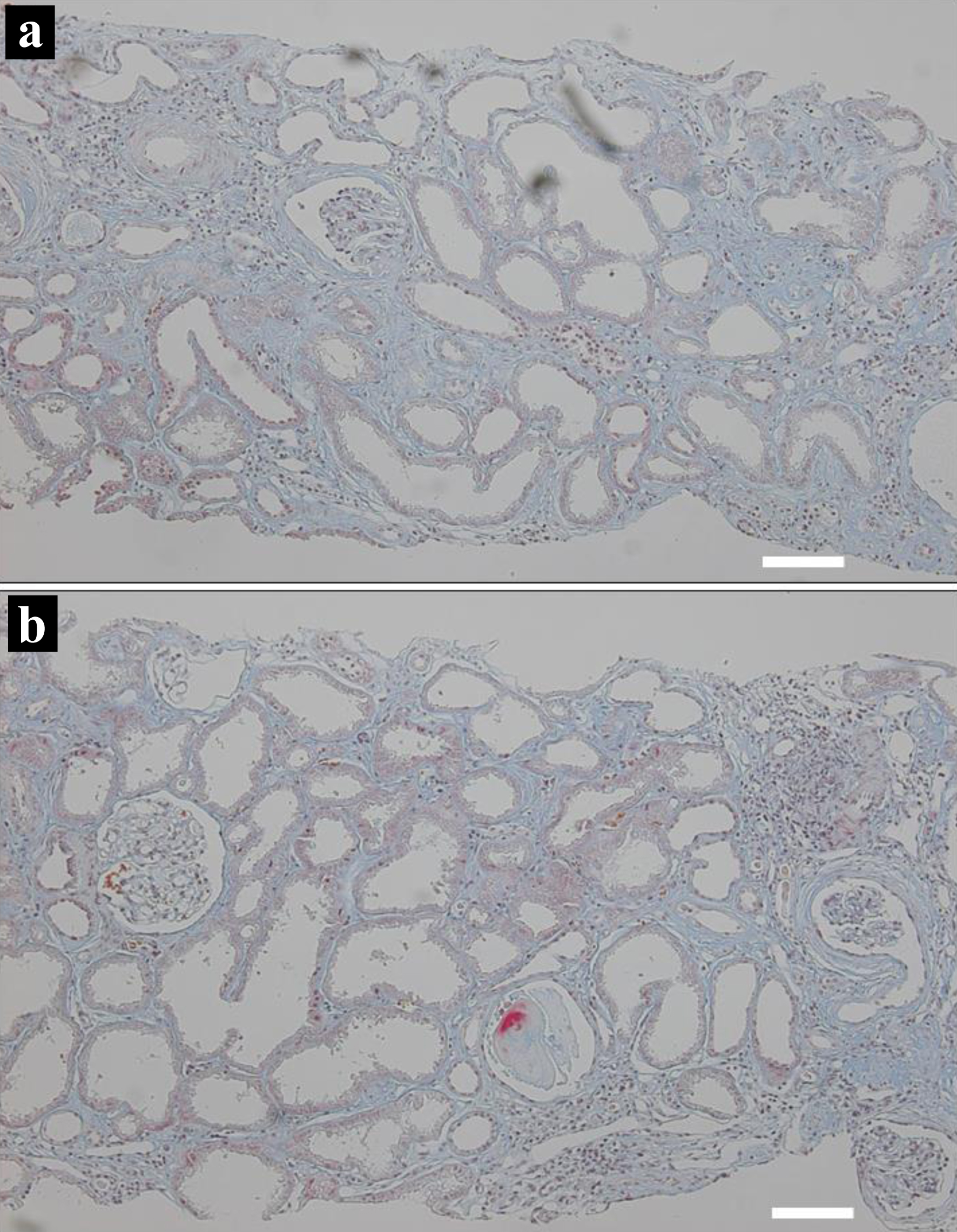 Click for large image |
Figure 1. Kidney biopsy at the age of 15 years. Two microscopic photographs (a, b) with color-faded Masson trichrome staining, showing diffuse interstitial fibrosis with lymphocytic infiltration, and renal tubular atrophy with irregular dilation. Scale bar = 100 µm. |
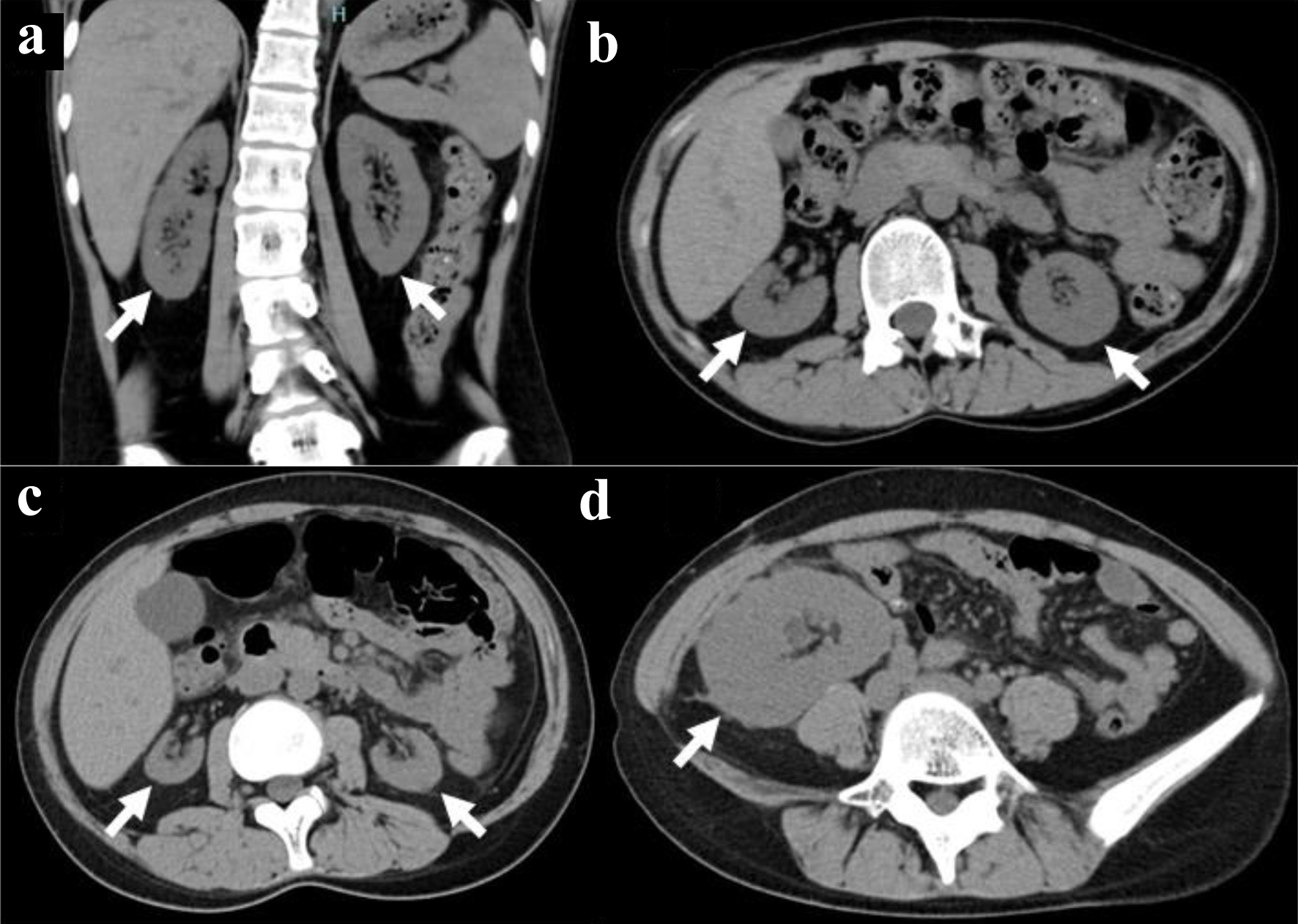
Three years later at the age of 18 years on the referral visit to the University Hospital, physical examinations, including neurological tests, showed nothing particular to be noted. She was taking oral prednisolone 5 mg daily and ferrous fumarate 100 mg daily. She did not smoke or drink alcohol. The height was 152.6 cm, the weight 38.3 kg, the blood pressure 135/79 mm Hg, and pulse rate 106/min. The 24-h urine volume was 2,200 mL and did not exceed a volume of 3,000 mL in repeat measurements later. In blood examinations (Table 1), serum creatine was high at 5.14 mg/dL (normal range, 0.46 - 0.79), blood urea nitrogen high at 51.5 mg/dL (normal range, 8.0 - 20.0), and estimated glomerular filtration rate (eGFR) low at 10.4 mL/min/1.73 mm2. Antinuclear antibody and anticardiolipin antibody were absent. Urinalysis was normal and did not detect proteinuria. Spot urine measurements showed an amount of total protein at 6.5 mg/dL and a high level of urine β2-microglobulin at 12.794 µg/mL (normal range, 0.0 - 0.289), while a normal level of urine N-acetylglucosaminidase (NAG) at 1.4 U/L (normal range, 0.3 - 11.5). Thoracic, abdominal, and pelvic computed tomography scans showed no abnormalities except for atrophic kidneys on both sides (Fig. 2a, b).
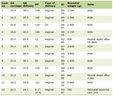 Click to view |
Table 1. Blood Examinations at the Initial
Visit and at the Latest Visit, 12 Years Later |
 Click for large image |
Figure 2. Computed tomography scans. Atrophic kidneys on both sides in coronal (arrows, a) and axial (arrows, b) images at the age of 18 years before kidney transplantation. Further atrophied kidneys (arrows, c) and kidney transplant in the normal dimension (arrow, d) at 19 years old, 8 months after kidney transplantation. |
At the age of 19 years, 4 months after the initial visit, she received an ABO-compatible living-donor kidney transplant from her 46-year-old father after three sessions of hemodialysis. At surgery, biopsy of the donor kidney showed two glomeruli with global sclerosis out of 24 glomeruli examined (Fig. 3a) in association with minimal interstitial fibrosis and lymphocytic infiltration. She began to have extended-release tacrolimus (tacrolimus ER) 4 mg daily and mycophenolate mofetil (MMF) 1,000 mg daily, 5 days before the kidney transplantation. One week after the kidney transplantation, oral prednisolone was increased to 20 mg daily and tacrolimus increased to 10 mg daily. The ureteral stent was removed 4 weeks after the kidney transplantation. According to the standard protocol for kidney transplantation, she underwent biopsy of the transplanted donor kidney to reveal no sign of rejection, no deposition of IgG, IgA, IgM, C3, C1q, C4d, or fibrin, 4 weeks and 14 months after the kidney transplantation. In the pathological findings, one glomerulus showed global sclerosis out of 13 glomeruli examined, in association with interstitial fibrosis and lymphocytic infiltration at 4 weeks (Fig. 3b), while 12 examined glomeruli were normal in association with interstitial fibrosis and lymphocytic infiltration as well as renal tubular atrophy at 14 months (Fig. 3c). Computed tomography scans in 8 months showed the kidney transplant in the normal dimension as well as her own kidneys in marked atrophy (Fig. 2c, d).
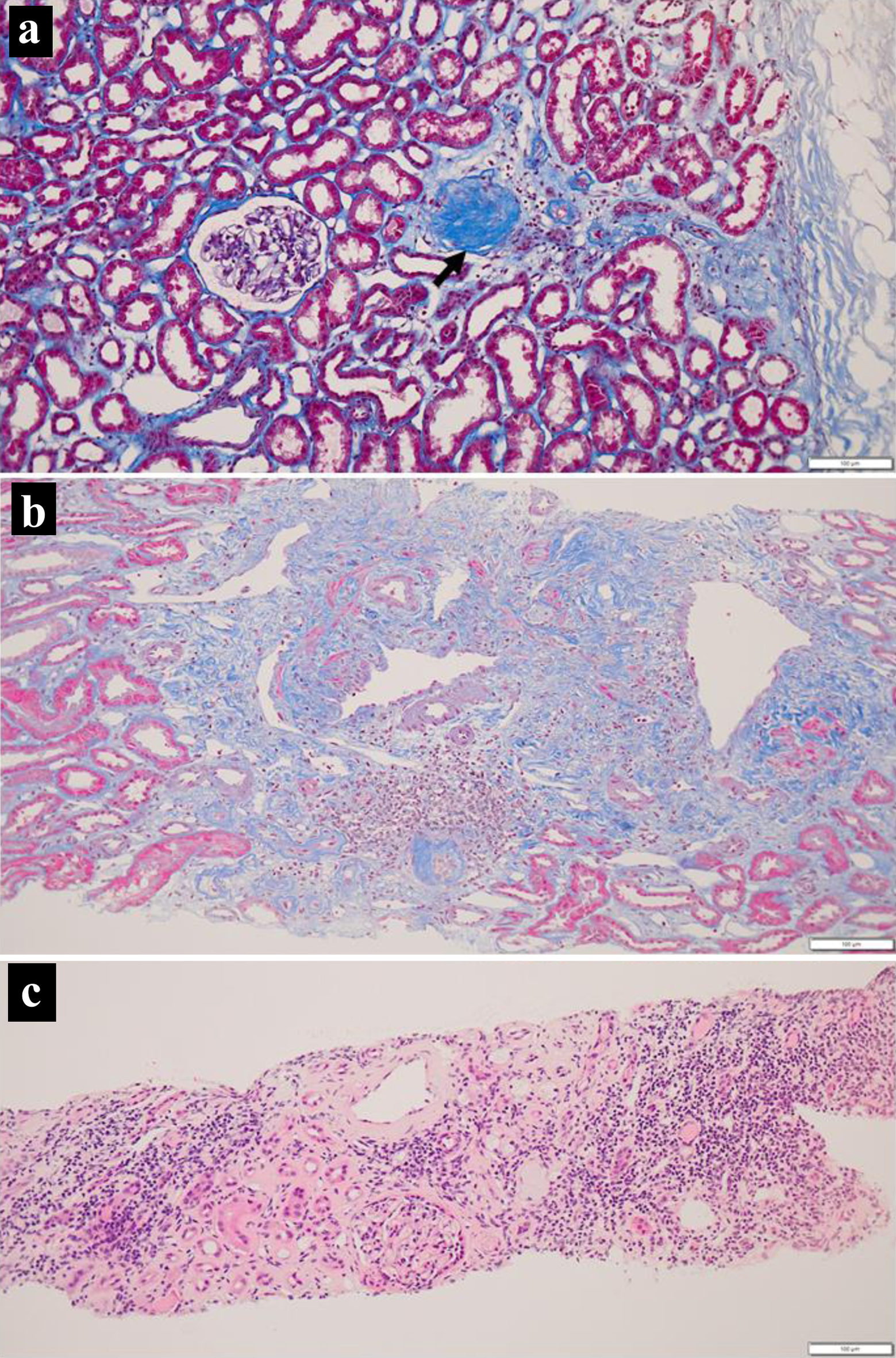 Click for large image |
Figure 3. Kidney biopsy at surgery (a), 4 weeks (b) and 14 months (c) after transplantation. Note one glomerulus with global sclerosis (arrow, a) and interstitial fibrosis with lymphocytic infiltration. Also note more advanced interstitial fibrosis with lymphocytic infiltration at 14 months (c). Masson trichrome stain in a and b, hematoxylin-eosin stain in c. Scale bar = 100 µm. |
At the age of 23 years, 4 years after the kidney transplantation, she noticed visual field defect for the first time, and visited a local eye doctor to be referred to the University Hospital. Specific questions revealed the history that she had noticed dark blindness in the early teens and that she had occasionally experienced difficulty in picking something up in late teens. The best-corrected visual acuity in decimals was 1.2 in both eyes, and the intraocular pressure was 15 mm Hg in both eyes. She had clear ocular media and showed mild diffuse non-pigmented retinal degeneration in the entire quadrants of the midperipheral fundus of both eyes outside the vascular arcade in the posterior pole with the normally appearing retina (Fig. 4a-d). In optical coherence tomography, the photoreceptor ellipsoid zone, which corresponded to the junction between the photoreceptor inner and outer segments, was preserved only in the limited area of the posterior pole of both eyes (Fig. 4e, f). Goldmann perimetry showed concentric visual field constriction to 5° meridians, only with the peripheral-rim field preservation in both eyes (Fig. 4g, h).
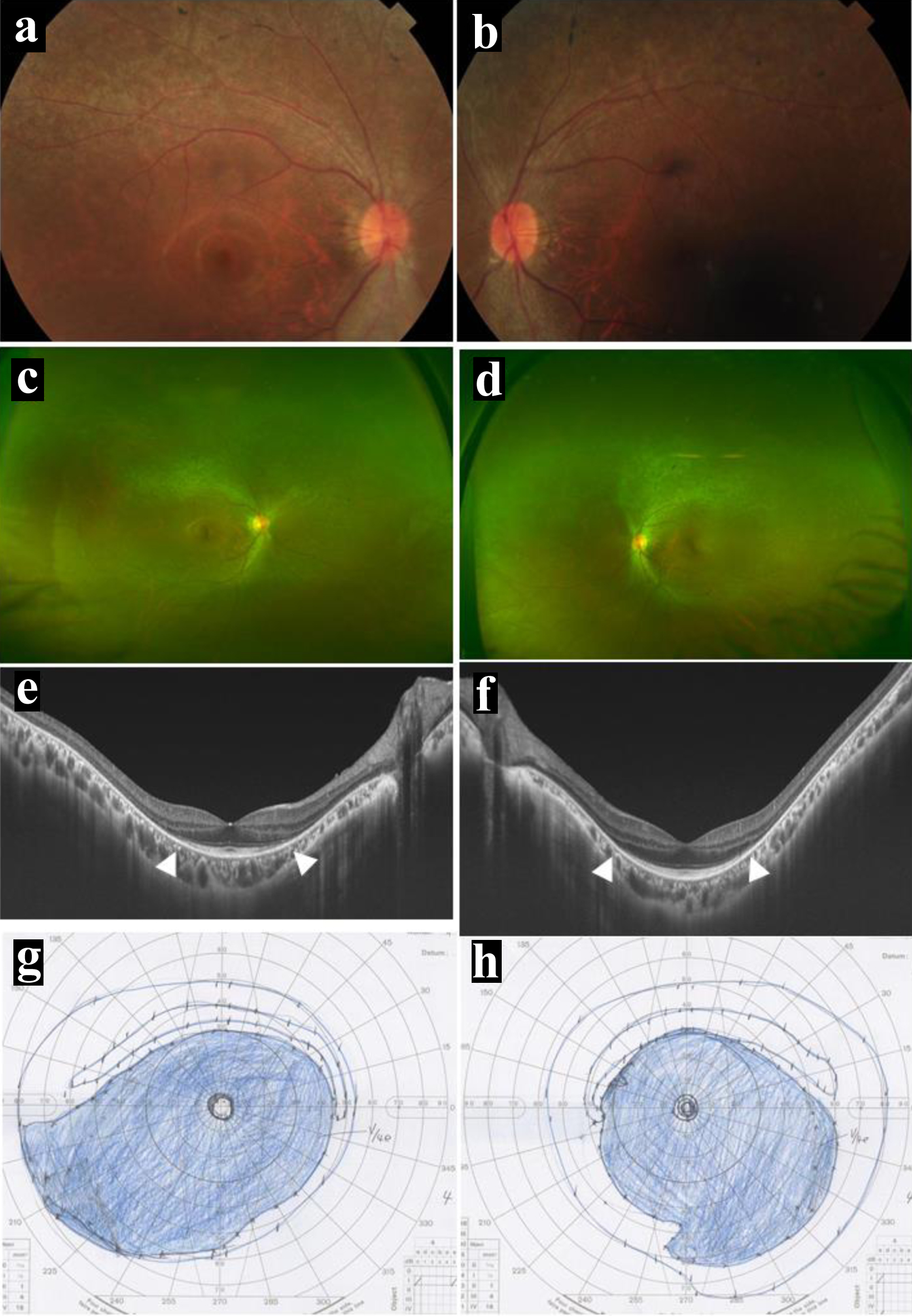 Click for large image |
Figure 4. Posterior pole (a in right eye, b in left eye) and wide-field (c in right eye, d in left eye) fundus photographs, and horizontal section images of optical coherence tomography (e in right eye, f in left eye) at the age of 23 years, 4 years after kidney transplantation. Note mild diffuse non-pigmented retinal degeneration outside the vascular arcade of the posterior pole (a-d) and the photoreceptor ellipsoid zones preserved only in the posterior pole (between arrowheads, e and f). Visual fields by Goldmann perimetry (g in left eye, h in right eye), showing concentric constriction to the central 5° meridians in both eyes, only with peripheral-rim fields left behind. |
She was followed up with basal doses of tacrolimus ER 5 mg daily, MMF 1,000 mg daily, and prednisolone 5 mg daily until the age of 31 years in 12 years after the kidney transplantation. The doses of tacrolimus ER and MMF had been decreased temporarily, based mainly on her gastrointestinal symptoms. Computed tomography scans showed the kidney transplant in the normal dimension as well as her own kidneys in extremely marked atrophy (Fig. 5a, b). The blood examinations showed creatinine at 0.94 mg/dL, blood urea nitrogen at 14.7 mg/dL, and eGFR at 57.3 mL/min/1.73 m2 (Table 1). In several years after the kidney transplantation, she had been psychologically unstable and had overcome the situation to work as a part-time government worker in the hometown. She maintained the normal retinal area in the posterior pole of both eyes (Fig. 5c-f), and had the same constricted visual field in both eyes as 8 years previously (Fig. 5g, h). She did not wish to have genetic testing.
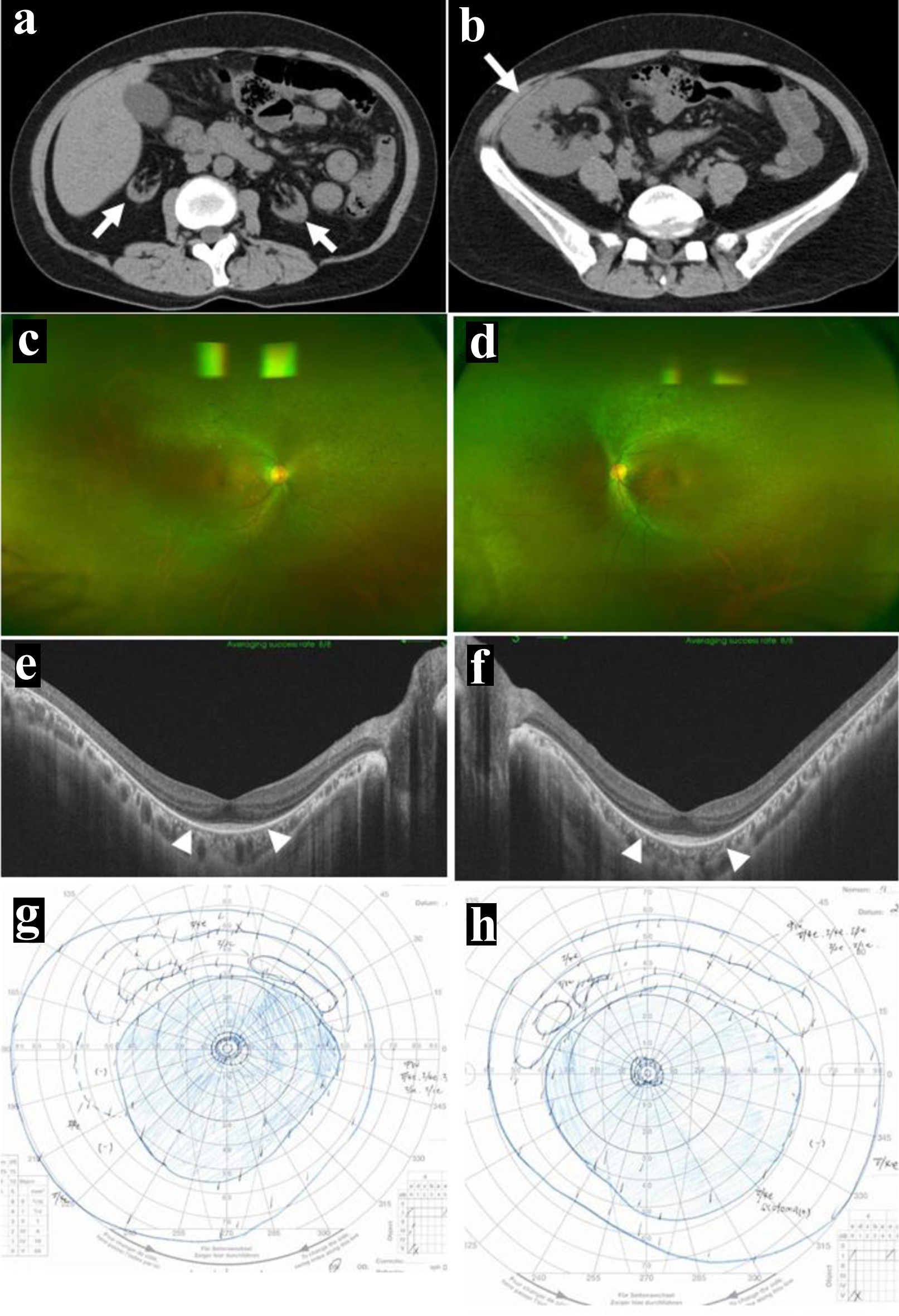 Click for large image |
Figure 5. At the age of 31 years, 12 years after kidney transplantation. Further atrophied kidneys (arrows, a) and kidney transplant in the normal dimension (arrow, b). Wide-field fundus photographs (c in right eye, d in left eye) and horizontal section images of optical coherence tomography (e in right eye, f in left eye), showing no change compared with 8 years previously (Fig. 4a-f). Note mild diffuse non-pigmented retinal degeneration outside the vascular arcade of the posterior pole (c, d) and the photoreceptor ellipsoid zones preserved only in the posterior pole (between arrowheads, e and f). Visual fields by Goldmann perimetry (g in left eye, h in right eye), showing concentric constriction to the central 5° meridians in both eyes, only with peripheral-rim fields left behind. |
| Discussion | ▴Top |
The present patient with no other physical or mental disorders developed end-stage renal disease in her teens. The pathological findings by the renal biopsy at the age of 15 years were global glomerulosclerosis, renal tubular dilation with irregular shape, and interstitial fibrosis with lymphocytic infiltration which are all consistent with the diagnosis of nephronophthisis in retrospect. As a preemptive therapy, she received a living-donor kidney transplant from her father at the age of 19 years. The course after the transplantation was stable in 12 years of follow-up.
It had passed 4 years since the renal transplantation when the patient was diagnosed with retinitis pigmentosa, based on typical ocular fundus findings in association with a symptom of night blindness. She had normal levels of visual acuity in both eyes but showed concentric visual field constriction to 5° meridian in both eyes. As the same as in the present patient, the diagnosis of retinitis pigmentosa would often be delayed later in the life of most patients since they maintain relatively better visual acuity enough for their daily living and working life. They would also be accustomed to narrow visual fields since the visual field would be deteriorating at a slow pace. Under the circumstances, patients with retinitis pigmentosa tend to be incidentally diagnosed as such when they visit eye doctors for other complaints.
In retrospect, the present patient had experienced night blindness and tended to bump often into something in her teens. With the diagnosis of retinitis pigmentosa, the diagnosis of Senior-Loken syndrome was thus reached in the present patient. Joubert syndrome and related disorders could be excluded by the absence of anomalies and the absence of neurological symptoms such as ataxia [5, 6]. Among the kidney transplant recipients, rare genetic diseases such as Senior-Loken syndrome would be a more common cause for end-stage renal disease than expected [9]. In a previous case report, as similar to the present patient, one patient with Senior-Loken syndrome was initially misdiagnosed as nephrosclerosis which was related to hypertensive disorders of pregnancy [10]. These previous reports have shown that it is not unusual for patients with Senior-Loken syndrome to follow a similar trajectory to the delayed diagnosis after kidney transplantation as a common real-world challenge. Genetic testing in patients with renal failure of unknown causes would help establish the diagnosis in the earlier phase [5, 8, 11].
The prognosis for kidney transplantation in patients with juvenile nephronophthisis was reported to be favorable even in living-donor transplant recipients [12]. Hereditary kidney diseases in autosomal recessive trait such as nephronophthisis are not the contraindication to living-donor transplantation from the families [13]. It is the standard protocol to check by biopsy a pathological sign of rejection in the transplanted donor kidney, 1 month and 1 year after the kidney transplantation. It should be noted in the present patient to observe the limited extent of global glomerulosclerosis and interstitial fibrosis with lymphocytic infiltration in specimens of the donor kidney from her father. A limited number of global glomerulosclerosis in the donor kidney would be the manifestation of the normal aging process in the 46-year-old father [14]. In contrast, lymphocytic infiltration with interstitial fibrosis is against the typical scene of rejection which shows the infiltration of lymphocytes around tubular epitheliums with HLA class II expression. Under the circumstances, global glomerulosclerosis with interstitial fibrosis and lymphocytic infiltration in the donor kidney might be a milder presentation of nephronophthisis with recessive traits [5, 6]. This reasoning remains speculative since genetic testing was not done in the patient and the parents, based on their wishes.
The present patient had been psychologically unstable especially after the kidney transplantation. It is natural to have a psychological overload in this patient who had suffered from renal failure in her early teens, received the living-donor renal transplant from her father, and was taking immunosuppressive drugs. Furthermore, she was diagnosed as retinitis pigmentosa in the course of the immunosuppressive treatment. She became mentally stable after she decided to move back to her hometown and started a part-time job as a government worker in the framework for persons with disabilities. In the ophthalmological follow-up, she was relieved to know no deterioration of visual acuity and visual fields in both eyes. The clinical situation in this patient emphasizes that psychological care and support are crucial in both medical and social contexts [15-17].
Conclusions
This report described a 19-year-old woman with end-stage renal disease who received a living-donor kidney transplant from her father as a preemptive therapy. At 15 years old, she was first pointed out to have poor renal function and underwent renal biopsy to show global glomerulosclerosis, renal tubular dilation in irregular shape, and marked interstitial fibrosis with lymphocytic infiltration. The protocol biopsy of the living-donor kidney from her father at renal transplantation and afterwards repeatedly showed a limited number of global glomerulosclerosis and interstitial fibrosis with lymphocytic infiltration. At the age of 23 years, 4 years after the kidney transplantation, she was, for the first time, diagnosed with retinitis pigmentosa, and hence, Senior-Loken syndrome. She was followed in the stable condition of the renal function as well as the visual acuity and visual fields in both eyes up until now in 12 years after the kidney transplantation. A limited number of global glomerulosclerosis and interstitial fibrosis with lymphocytic infiltration in the donor kidney from the father might be a milder presentation of the disease with recessive inheritance.
Learning points
Patients with retinitis pigmentosa usually have good visual acuity with the central visual fields which are preserved in both eyes and tend not to notice by themselves the narrowing of visual fields since the disease is slow in deterioration. Specific questions are useful to detect night blindness and narrow visual fields such as bumping into something in the dark.
Acknowledgments
None to declare.
Financial Disclosure
The authors receive no financial support for the research, authorship, and/or publication of this article.
Conflict of Interest
The authors declare no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Informed Consent
Written informed consent was obtained from the patient for her anonymized information to be published in this article. Ethics committee review was not applicable to case reports, based on the Ethical Guidelines for Medical and Health Research Involving Human Subjects, issued by the Government of Japan.
Author Contributions
TM, as an ophthalmologist, and YO, HM, and JW, as nephrologists, followed and treated the patient, TT, as a pathologist, made the pathological diagnosis, MA, as a urologist, did transplantation surgery and followed the patient. TM wrote the manuscript, and YO, HM, JW, TT, and MA did critical review of the manuscript, and all authors approved the final version of the manuscript.
Data Availability
The data supporting the findings of this study are available from the corresponding author upon reasonable request.
| References | ▴Top |
- Verbakel SK, van Huet RAC, Boon CJF, den Hollander AI, Collin RWJ,
Klaver CCW, Hoyng CB, et al. Non-syndromic retinitis pigmentosa. Prog Retin Eye Res.
2018;66:157-186.
doi pubmed - Karuntu JS, Almushattat H, Nguyen XT, Plomp AS, Wanders RJA, Hoyng
CB, van Schooneveld MJ, et al. Syndromic retinitis pigmentosa. Prog Retin Eye Res.
2025;107:101324.
doi pubmed - McConnachie DJ, Stow JL, Mallett AJ. Ciliopathies and the kidney: a
review. Am J Kidney Dis. 2021;77(3):410-419.
doi pubmed - Matsuo T, Tanaka T, Tsuji K. Presumed choroidopathy of IgG4-related
disease discovered during 16-year follow-up of a patient with polycystic kidney disease. Cureus.
2024;16(10):e70865.
doi pubmed - Wolf MTF, Bonsib SM, Larsen CP, Hildebrandt F. Nephronophthisis: a
pathological and genetic perspective. Pediatr Nephrol. 2024;39(7):1977-2000.
doi pubmed - Hildebrandt F, Attanasio M, Otto E. Nephronophthisis: disease
mechanisms of a ciliopathy. J Am Soc Nephrol. 2009;20(1):23-35.
doi pubmed - Ronquillo CC, Bernstein PS, Baehr W. Senior-Loken syndrome: a
syndromic form of retinal dystrophy associated with nephronophthisis. Vision Res.
2012;75:88-97.
doi pubmed - Yahalom C, Volovelsky O, Macarov M, Altalbishi A, Alsweiti Y,
Schneider N, Hanany M, et al. Senior-Loken Syndrome: a case series and review of the Renoretinal
phenotype and advances of molecular diagnosis. Retina. 2021;41(10):2179-2187.
doi pubmed - Quaglia M, Musetti C, Ghiggeri GM, Fogazzi GB, Settanni F, Boldorini
RL, Lazzarich E, et al. Unexpectedly high prevalence of rare genetic disorders in kidney
transplant recipients with an unknown causal nephropathy. Clin Transplant.
2014;28(9):995-1003.
doi pubmed - Hirai Y, Mizumoto A, Mitsumoto K, Uzu T. Senior-Loken syndrome
misdiagnosed as nephrosclerosis related to hypertensive disorders of pregnancy. BMJ Case Rep.
2020;13(10):e236137.
doi pubmed - Wang J, Li S, Jiang Y, Wang Y, Ouyang J, Yi Z, Sun W, et al.
Pathogenic variants in CEP290 or IQCB1 cause earlier-onset retinopathy in
Senior-Loken syndrome compared to those in INVS, NPHP3, or NPHP4.
Am J Ophthalmol. 2023;252:188-204.
doi pubmed - Avci B, Baskin E, Gulleroglu K, Caltik Yilmaz A, Kantar A, Akdur A,
Moray G, et al. Long-term outcomes of kidney transplant recipients with juvenile
nephronophthisis. Exp Clin Transplant. 2022;20(Suppl 3):122-125.
doi pubmed - Vnucak M, Granak K, Skalova P, Laca L, Mokan M, Dedinska I.
Living-related kidney transplantation in a patient with juvenile nephronophthisis. Nephron.
2020;144(11):583-588.
doi pubmed - Kremers WK, Denic A, Lieske JC, Alexander MP, Kaushik V, Elsherbiny
HE, Chakkera HA, et al. Distinguishing age-related from disease-related glomerulosclerosis on
kidney biopsy: the Aging Kidney Anatomy study. Nephrol Dial Transplant.
2015;30(12):2034-2039.
doi pubmed - Akyirem S, Forbes A, Wad JL, Due-Christensen M. Psychosocial
interventions for adults with newly diagnosed chronic disease: A systematic review.
J Health Psychol. 2022;27(7):1753-1782.
doi pubmed - Hu N, Wang A, Chang T. Social support mediates the relationship
between illness perception and psychosocial adaptation among young and middle-aged kidney
transplant recipients in China. Front Psychol. 2023;14:1062337.
doi pubmed - Wube TB, Asgedom SG, Mengesha AG, Bekele YA, Gebrekirstos LG. Behind
the healing: exploring the psychological battles of kidney transplant patients: a qualitative
insight. Health Sci Rep. 2025;8(2):e70511.
doi pubmed
This
article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0
International License, which permits unrestricted non-commercial use, distribution, and
reproduction in any medium, provided the original work is properly cited.
Journal
of Medical Cases is published by Elmer Press Inc.