| Journal of Medical Cases, ISSN 1923-4155 print, 1923-4163 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Med Cases and Elmer Press Inc |
| Journal website https://jmc.elmerpub.com |
Case Report
Volume 16, Number 2, February 2025, pages 48-54

An Unusual Case of Extracavitary/Solid Variant Primary Effusion Lymphoma With Associated Hemophagocytic Lymphohistiocytosis
Chika Iguha, b, Julie Kima, Akudo Akaraonyea, Amani Minjaa, Xin Qinga
aDepartment of Pathology and Laboratory Medicine, Harbor-UCLA Medical Center,
Torrance, CA 90502, USA
bCorresponding Author: Chika Iguh, Department of
Pathology and Laboratory Medicine, Harbor-UCLA Medical Center, Torrance, CA 90502, USA
Manuscript submitted November 13, 2024, accepted December 19, 2024, published online January 9,
2025
Short title: Extracavitary PEL With HLH
doi: https://doi.org/10.14740/jmc5084
| Abstract | ▴Top |
Primary effusion lymphoma (PEL) is a rare, aggressive large B-cell lymphoma variant that is invariably associated with human herpesvirus 8 (HHV8), predominantly in human immunodeficiency virus (HIV)-infected patients, and its oncogenicity is often augmented by coinfection with Epstein-Barr virus. It typically presents as a serous effusion in body cavities without detectable solid tumors. The extracavitary variant of PEL may represent a diagnostic challenge. A 37-year-old man with HIV/acquired immunodeficiency syndrome (AIDS) was transferred to our hospital for evaluation of a mediastinal mass with associated clinically diagnosed hemophagocytic lymphohistiocytosis (HLH), fever, pancytopenia, hepatosplenomegaly, retroperitoneal lymphadenopathy, and wasting syndrome. Contrast-enhanced computed tomography showed a large soft tissue mass extending along the middle/posterior mediastinum into the left hilum and a large left pleural effusion. Endoscopic fine-needle biopsy of the lesion showed sheets of large pleomorphic lymphoma cells with prominent nucleoli and abundant cytoplasm. These cells were also seen on the cytospin smear of pleural fluid. Immunohistochemical stains showed lymphoma cells positive for CD3 (small subset), CD45, CD138, MUM-1, and HHV8 and negative for CD5, CD20, CD30, ALK1, AE1/3, and PAX-5. The lymphoma cells were also positive for Epstein-Barr virus-encoded RNA (EBER) (in situ hybridization). Solid masses in extracavitary PEL have been shown to involve lymph nodes and/or solid organs such as the gastrointestinal tract, lung, liver, spleen, and skin, with a similar phenotype as classic PEL except that they may express B-cell markers with lower expression of CD45 and/or aberrant coexpression of T-cell antigens. This case illustrates the unusual manifestation of PEL as a mediastinal mass with associated HLH.
Keywords: Primary effusion lymphoma; Extracavitary primary effusion lymphoma; Solid variant; Mediastinal; Hemophagocytic lymphohistiocytosis
| Introduction | ▴Top |
Primary effusion lymphoma (PEL) is a rare and aggressive subtype of B-cell non-Hodgkin lymphoma, strongly associated with Kaposi’s sarcoma-associated herpesvirus (KSHV, also known as human herpesvirus 8 (HHV-8)) and frequent co-infection with Epstein-Barr virus (EBV) [1, 2]. It predominantly affects immunocompromised patients, especially those with advanced human immunodeficiency virus (HIV)/acquired immunodeficiency syndrome (AIDS) and is recognized as an AIDS-defining malignancy [3]. While classic PEL typically presents as an effusion without a detectable mass, the extracavitary variant (extracavitary primary effusion lymphoma (EC-PEL)) manifests as a solid tumor, often complicating its diagnosis [4]. The prognosis of PEL is generally poor, with median survival times of less than a year despite aggressive therapy.
The coexistence of PEL with hemophagocytic lymphohistiocytosis (HLH) further exacerbates the clinical course. HLH is a life-threatening hyper-inflammatory syndrome triggered by dysregulated immune responses, frequently observed in association with hematologic malignancies, although its association with PEL is rarely reported in the literature. This case report presents a highly unusual instance of EC-PEL involving the middle and posterior mediastinum, accompanied by HLH in a patient with HIV/AIDS. The case underscores the diagnostic and therapeutic challenges posed by EC-PEL in the context of profound immunosuppression, compounded by the presence of HLH and disseminated opportunistic infections, highlighting the need for early recognition and multi-faceted treatment approaches.
| Case Report | ▴Top |
Investigations
The patient is a 37-year-old male with a history of HIV/AIDS with questionable antiretroviral therapy (ART) adherence, methamphetamine use disorder and wasting syndrome. He initially presented to an outside hospital with symptoms of small bowel obstruction and was subsequently admitted due to spiking fevers. His history at the outside hospital included normocytic anemia, marked thrombocytopenia, a presenting CD4 count of less than 20 cells/µL (4%), and a viral load of 4,670,000 copies/mL. Bone marrow aspiration and biopsy performed at the outside hospital revealed a hypercellular bone marrow with erythroid hypoplasia and reactive plasmacytosis. There was no evidence of increased blasts, lymphoma, or granuloma; and there was no mention of the presence of hemophagocytosis or macrophages with or without cytoplasmic vacuoles. Acid-fast stain and Grocott-Gomori’s methenamine silver stain were reported as negative for acid-fast bacilli and fungal organisms, respectively.
Upon admission to our hospital, the patient reported worsening bilateral lower extremity swelling, shortness of breath with exertion, decreased appetite, dysphagia, weight loss, and blurry vision. His vital signs revealed borderline tachycardia (90 beats per minute (bpm)).
Diagnosis
Laboratory findings and physical examination
On admission, the patient’s labs revealed worsening pancytopenia, with a hemoglobin of 6.6 g/dL following one unit of red blood cell (RBC) transfusion, white blood cell count of 1.0 × 109/L, and platelet count of 37 × 109/L. He also exhibited hyponatremia (124 mEq/L), severe hypoalbuminemia, and elevated liver function tests. A urine toxicology screen was positive for amphetamines. On physical examination, the patient appeared thin, with a faint systolic murmur most prominent at the left lower sternal border and 1+ to 2+ pitting edema over the anterior shins.
Imaging studies
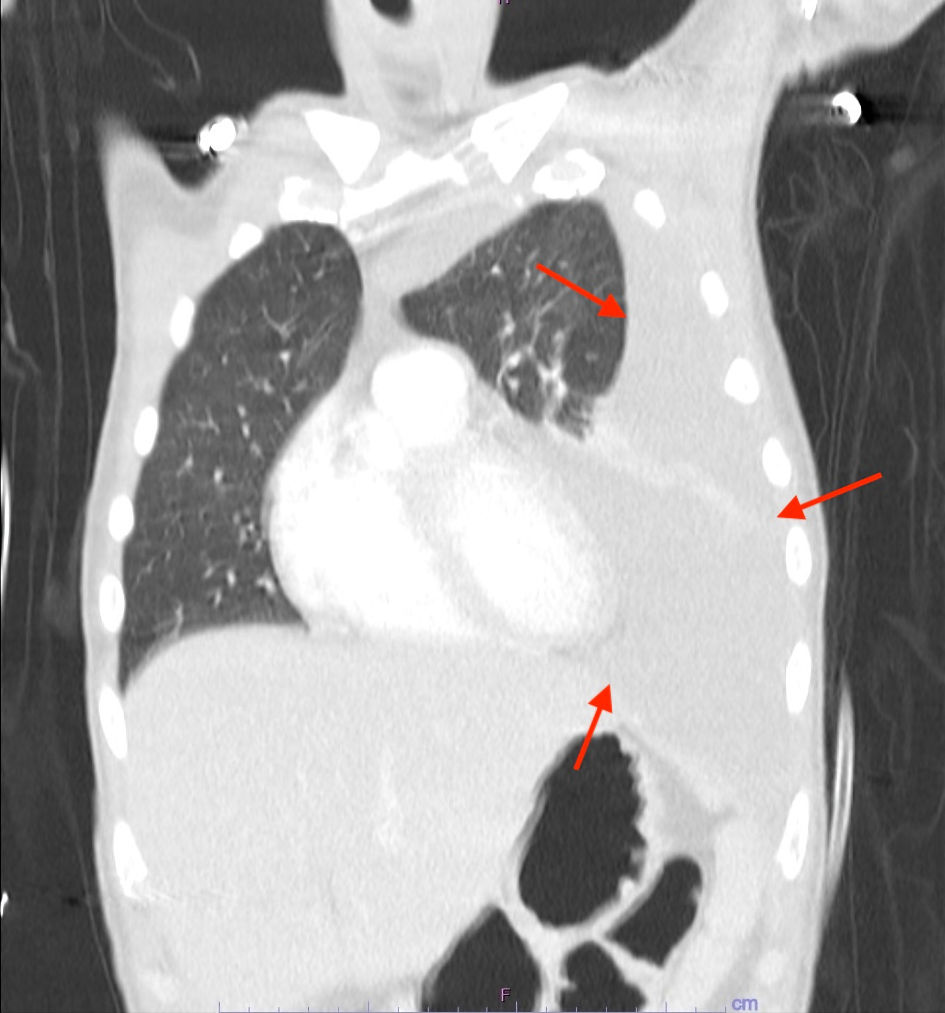
Computed tomography (CT) of the thorax with contrast revealed an 8.7 × 7.1 × 4 cm soft tissue mass extending along the midline and posterior mediastinum into the left hilum (Fig. 1). This lesion exerted a mass effect on the left atrium and encased several pulmonary veins, resulting in mild narrowing of the right inferior pulmonary vein, severe narrowing of the left superior pulmonary vein, and near-complete occlusion of the left inferior pulmonary vein (Fig. 1). There was also effacement of the fat plane along the anterior esophagus with esophageal wall thickening, raising concerns for possible involvement (Fig. 1). Additionally, there was a small left pleural effusion, which eventually developed into a large pleural effusion occupying the majority of the left hemithorax, with loculated fluid along the left anteromedial hemithorax (Fig. 2).
 Click for large image |
Figure 1. CT of the chest with contrast demonstrating an 8.7 × 7.1 × 4 soft tissue mass extending along the middle and posterior mediastinum into the left hilum (arrow). The lesion is exerting mass effect on the left atrium, encasing several pulmonary veins, and effacing the fat plane along the anterior esophagus with esophageal wall thickening. CT: computed tomography. |
 Click for large image |
Figure 2. CT thorax with contrast demonstrating a large pleural effusion occupying the majority of the left hemithorax (arrows). CT: computed tomography. |
CT abdomen/pelvis showed hepatosplenomegaly, lymphadenopathy throughout the retroperitoneum and mesentery (up to 9 mm in short axis diameter), and a moderate amount of free fluid in the pelvis. Given the patient’s HIV status and the extent of the mediastinal mass, an extensive infection was considered a possible differential diagnosis in addition to malignancy.
Pathology results
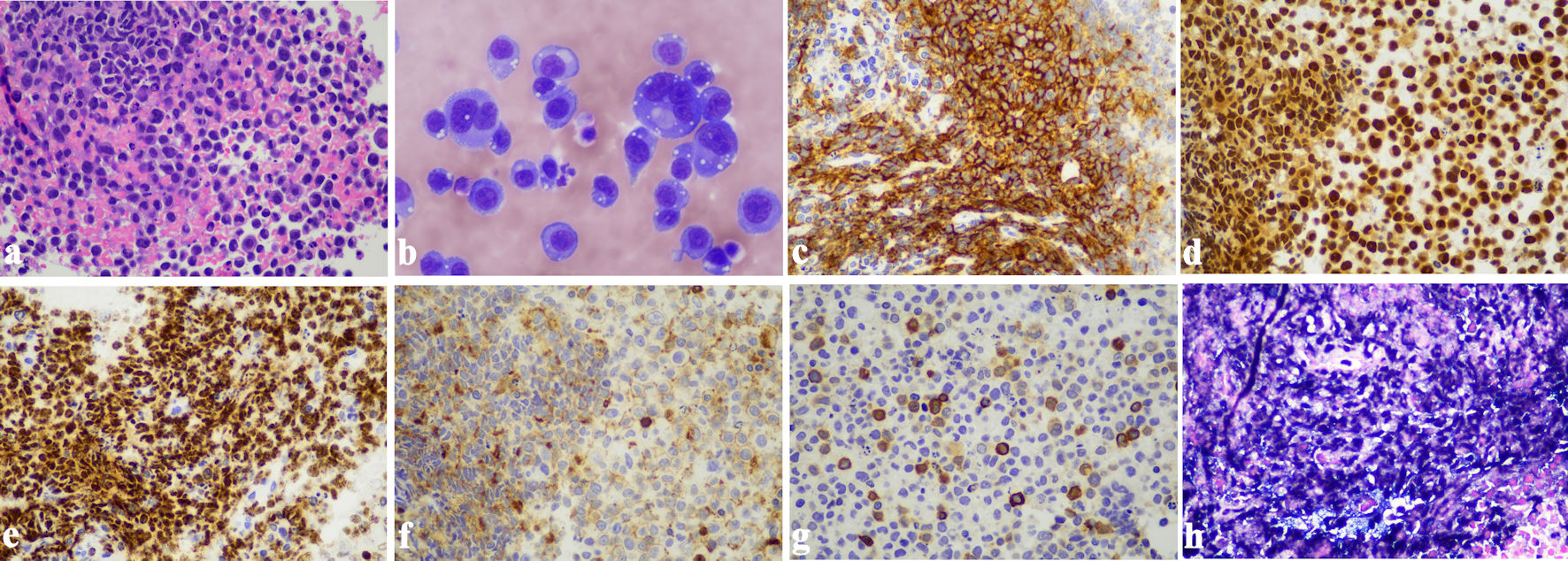
Endoscopic ultrasound-guided fine needle biopsy of the mediastinal mass revealed sheets of large pleomorphic lymphoma cells with prominent nucleoli and abundant cytoplasm (Fig. 3a). Immunohistochemical (IHC) stains showed the lymphoma cells were positive for CD138 (Fig. 3c), multiple myeloma oncogene 1 (MUM-1) (Fig. 3d), and HHV8 (Fig. 3e), weakly positive for CD45 (Fig. 3f), with a small subset positive for CD3 (Fig. 3g). Ki67 IHC staining indicated a high proliferation rate (not shown). Additionally, Epstein-Barr virus-encoded RNA (EBER) in situ hybridization (ISH) was positive (Fig. 3h). The cells were negative for CD5, CD20, CD30, ALK1, pan-cytokeratin AE1/AE3, and PAX-5 (not shown). A diagnosis of EC-PEL was made. Later, a cytospin smear of his pleural fluid also demonstrated large lymphoma cells with marked pleomorphism, prominent nucleoli, and deep blue plasmacytoid cytoplasm (Fig. 3b), consistent with the diagnosis of PEL. There was no evidence of multicentric Castleman disease.
 Click for large image |
Figure 3. Primary effusion lymphoma. (a) Sheets of large pleomorphic lymphoma cells with prominent nucleoli and abundant cytoplasm, morphologically consistent with EC-PEL (H&E stain, original magnification: × 400). (b) Cytospin smear of pleural fluid showing PEL (Wright-Giemsa stain, original magnification: × 600). (c) Strong positive immunostaining for CD138 in neoplastic cells (immunohistochemistry, original magnification: × 400). (d) Strong positive immunostaining for MUM-1 in neoplastic cells (immunohistochemistry, original magnification: × 400). (e) The lymphoma cells demonstrate strong positive immunostaining for HHV8 (immunohistochemistry, original magnification: × 400). (f) Weak positive immunostaining for CD45 in neoplastic cells (immunohistochemistry, original magnification: × 400). (g) A small subset of neoplastic cells showed positive immunostaining for CD3 (immunohistochemistry, original magnification: × 400). (h) In situ hybridization for EBER was positive in the lymphoma cells (original magnification: × 400). EBER: Epstein-Barr virus-encoded RNA; EC-PEL: extracavitary primary effusion lymphoma; H&E: hematoxylin and eosin stain; PEL: primary effusion lymphoma. |
Treatment and hospital course
Several weeks into hospital admission, the patient was clinically diagnosed with concomitant HLH, based on fulfillment of at least five HLH-2004 criteria and an HScore of 236 (Tables 1, 2). Because of his worsening and unstable condition and already meeting criteria for HLH without histology, repeat bone marrow biopsy was foregone.
 Click to view |
Table 1. Patient HScore |
Click to view |
Table 2. HLH-2004 Criteria in Present
Case |
The patient underwent five cycles of chemotherapy (one cycle of CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisone), followed by two cycles of EPOCH (etoposide, prednisone, vincristine, cyclophosphamide, and doxorubicin) and three cycles of V-EPOCH) and received appropriate combination therapy for HIV, Pneumocystis jiroveci pneumonia prophylaxis, and HLH (etoposide 150 mg/m2 intravenous (IV) twice per week, tapering to once per week after 2 weeks, and high-dose dexamethasone with an 8-week taper).
His hospital course was complicated by disseminated Mycobacterium avium complex (MAC) infection. The duodenal thickening seen on upper endoscopy raised concerns for gastrointestinal involvement of MAC, which was confirmed on jejunal biopsy. Furthermore, brain magnetic resonance imaging after three cycles of chemotherapy revealed multiple scattered parenchymal lesions with rim enhancement, raising concerns for either primary central nervous system (CNS) lymphoma, CNS involvement by PEL or infectious etiologies (i.e., CNS toxoplasmosis, granulomatous disease, fungal infection, or tuberculosis). Cerebrospinal fluid analysis detected the presence of EBV DNA, but toxoplasmosis serology was negative. A definitive diagnosis of the brain lesions was precluded by medical decision to not biopsy due to deemed high risk in the setting of very poor patient condition.
The patient’s sixth cycle of chemotherapy was held due to the severity of his disseminated MAC with gastrointestinal involvement. He eventually refused further chemotherapy and opted for palliative care. As his condition deteriorated, he became altered, nonresponsive, and noncommunicative. His surrogate decision-maker agreed to transition to comfort care, and the patient ultimately was declared deceased in a comfort care room soon thereafter.
| Discussion | ▴Top |
Overview of EC-PEL
PEL, originally described by Cesarman et al in 1995, is a rare and aggressive subtype of large B-cell lymphoma strongly associated with HHV-8, also known as KSHV [5]. Extracavitary/solid-variant PEL (EC-PEL) was first described by Chadburn et al in 2004 [6]. While classic PEL typically presents as a lymphomatous effusion within body cavities such as the pleura, pericardium, or peritoneum, EC-PEL presents as a solid tumor mass with or without accompanying effusion, posing a significant diagnostic challenge [4, 7].
Epidemiology and clinical presentation
PEL predominantly occurs in the context of immunosuppression, most notably in HIV-positive individuals, where it serves as an AIDS-defining illness [5]; it has also been documented in the posttransplant setting [8]. This lymphoma subtype represents 3-4% of all HIV-associated non-Hodgkin lymphomas (NHLs), less than 1% of all lymphomas and typically shows an aggressive nature with poor prognosis [1, 4, 9]. EC-PEL, while rarer, has been reported in various extranodal sites, including the gastrointestinal tract, lungs, skin, and CNS, among others [10, 11]. The present case is particularly unusual due to extranodal and extracardiac involvement of EC-PEL in the middle and posterior mediastinum, a location less frequently documented in the literature [7, 10]. Katz et al (2015) reported the first case of EC-PEL presenting as a right paracardial mass [7]. However, since then, to the best of our knowledge, no additional reports of extranodal EC-PEL involving the middle mediastinal region have been reported. Furthermore, extensive extranodal and extracardiac involvement of the middle mediastinum with posterior mediastinal extension, encasing and involving the pulmonary vessels and esophageal wall, as demonstrated by the present case, is a unique clinical presentation. In this regard, the present case adds to the growing body of literature characterizing EC-PEL.
Pathogenesis
The pathogenesis of PEL is intricately linked to HHV-8: HHV-8 is detected in nearly all cases of PEL, making its identification a key diagnostic feature [12, 13]. The virus encodes multiple proteins, including viral cyclin (v-cyclin), latency-associated nuclear antigen (LANA), viral caspase-8, FLICE-like inhibitory protein (vFLIP), virally encoded cytokines (i.e., viral interleukin-6 (vIL-6)), viral chemokines (i.e., vCCL1, vCCL2, v CCL3), CD200, and interferon (IFN)-regulatory factors (i.e., vIRF1, vIRF2, vIRF3, and vIRF4), among others [14]. Human interleukin-6 (IL-6) and IL-10 have also been shown to be upregulated [15]. These proteins contribute to cell proliferation, immune evasion, and inhibition of apoptosis during the latency and lytic reactivation phases and are crucial in driving the oncogenic process. Additionally, EBV co-infection, which is observed in approximately 50-80% of PEL cases, may also contribute to the oncogenic process, although its pathogenic role remains less clear [9]. In EC-PEL, the solid tumor mass represents a localized form of the disease, potentially indicating a variation in the pathophysiologic mechanisms observed in classic PEL. However, the underlying oncogenic role of HHV-8 remains consistent across both forms.
Morphologic and immunophenotypic distinctions in PEL
PEL is morphologically characterized by the presence of large, pleomorphic cells that may display immunoblastic, plasmablastic, or anaplastic features. These neoplastic cells often show abundant basophilic cytoplasm, eccentric nuclei, and prominent nucleoli, with high mitotic activity and the presence of apoptotic bodies [1, 9]. In effusion specimens, the lymphoma cells are typically discohesive and exhibit irregular nuclear contours, often mimicking other malignancies such as other large cell lymphomas and poorly differentiated carcinomas [4, 14].
Immunophenotypically, PEL is notable for its lack of pan-B-cell marker expression, including CD19, CD20, and PAX5 [1, 16]. Instead, PEL commonly expresses plasma cell markers such as CD38 and CD138, in addition to MUM1 and CD30. As aforementioned, these cells are also positive for HHV-8, and in situ hybridization is often positive for EBV [14]. The Ki-67 proliferation index is generally high, reflecting the aggressive nature of this entity [1].
The distinction between classic PEL and EC-PEL regarding their morphologic and immunophenotypic profiles is not very significant. The landmark study by Chadburn et al [6] showed that classic and EC-PEL showed overlapping features, including HHV-8 positivity, frequent EBV co-infection, and similar immunophenotypes. However, despite both forms being associated with HHV-8, EC-PEL has been shown in some studies to exhibit a higher frequency of aberrant T-cell marker expression, such as CD3, as demonstrated in the present case [17]. This variation can be seen in up to 30% of all EC-PEL cases and should not complicate the diagnosis or lead to misclassification as a T-cell lymphoma [1, 16, 18]. Additionally, in a study of 70 PEL cases, Hu et al found that EC-PEL cases without effusion tended to infrequently express MUM1, CD45, CD30 and epithelial membrane antigen (EMA), when compared to classic PEL and cases with both effusion and extracavitary involvement [17].
Morphologic and immunophenotypic overlap with other aggressive lymphomas, particularly plasmablastic lymphoma, ALK-positive large B-cell lymphoma, anaplastic plasmacytoma, and HHV8-positive large B-cell lymphoma, may challenge accurate diagnosis; the combination of positive CD138 and HHV8 and negative B-cell markers will aid the pathologist when the diagnosis is unclear.
HLH association
HLH is a life-threatening hyper-inflammatory syndrome characterized by excessive and dysregulated immune activation, leading to uncontrolled cytokine release and resulting in systemic inflammation and multi-organ dysfunction. HLH is broadly classified as either primary (familial), which is associated with genetic mutations affecting immune regulation, or secondary (acquired), which is often triggered by infections, hematologic malignancies, or autoimmune disorders. The present case represents one of very few cases of acquired HLH secondary to PEL reported in the literature [19-21].
The HLH-2004 diagnostic criteria remain the gold standard for diagnosing HLH, requiring the fulfillment of at least five of eight clinical and laboratory parameters: persistent fever, splenomegaly, cytopenia affecting two or more hematopoietic cell lines, hypertriglyceridemia, hypofibrinogenemia, hemophagocytosis in bone marrow or other organs, reduced or absent natural killer (NK) cell activity, and elevated ferritin levels (> 500 ng/mL) [22]. Additional supportive findings may include elevated soluble interleukin-2 receptor (sIL-2R) levels, which reflect excessive T-cell activation [22, 23]. Hemophagocytosis contributes directly to the cytopenias observed in HLH, as well as other laboratory abnormalities such as hyperferritinemia and hypertriglyceridemia, but its presence on histology is not required to make the diagnosis [22, 24, 25]. The excessive macrophage activation, along with continuous release of pro-inflammatory cytokines, such as IFN-gamma and tumor necrosis factor-alpha, perpetuates the inflammatory cascade.
In patients with PEL, particularly those who are immunocompromised, the simultaneous presence of HLH requires a high degree of clinical suspicion to avoid diagnostic delays, as these conditions can exacerbate one another, and early treatment may improve prognosis [21, 26]. Biomarkers like elevated IL-6 and IL-10 have emerged as important indicators of prognosis in PEL, and their involvement in immune dysregulation suggests a potential mechanism linking PEL with HLH [15]. Unfortunately, these biomarkers were not measured in our patient. Management of PEL-associated HLH requires a multidisciplinary approach. The HLH-94 protocol, which includes immunosuppressive agents such as corticosteroids and etoposide, remains the cornerstone of HLH treatment. Addressing the primary malignancy is crucial for controlling the hyperinflammatory response. Treatment of PEL involves multi-agent chemotherapy, often including CHOP-based regimens, along with antiviral therapies.
In the present case, the patient was treated with V-EPOCH (vincristine (Oncovin), etoposide, prednisone, cyclophosphamide, and hydroxydaunorubicin (doxorubicin)), highly active ART (bictegravir, emtricitabine and tenofovir alafenamide), and immunosuppressive therapy with taper. However, he succumbed to disease progression approximately 5 months after diagnosis.
Prognosis
The prognosis for patients with PEL is generally poor, with median survival times ranging between 6 and 9 months, although those with EC-PEL may exhibit slightly improved survival outcomes [4, 27, 28]. In a comprehensive analysis of 51 HIV-associated PEL cases by Guillet et al, EC-PEL demonstrated better disease-free survival, as no relapses were reported in patients who achieved complete remission (CR); whereas classic PEL exhibited a relapse rate of 62% post-CR [29]. However, overall survival (OS) remained poor for both groups, with a median of 10 months [29]. Factors influencing prognosis include the extent of disease at diagnosis, the patient’s immune status, ART before PEL diagnosis, lactate dehydrogenase (LDH) levels and the presence of co-infections such as EBV or disseminated MAC [27]. A study of 20 patients by Lurain et al interestingly demonstrated a significant association between EBV-positive tumor status and improved survival in PEL [15]. The study also demonstrated a significant association between elevated IL-6 expression and inferior survival [15]. The number of involved body cavities and the response to initial therapy are also critical determinants of survival [13, 14, 27]. As such, the rapid progression observed in the present case despite aggressive intervention may be attributed to several factors: inconsistent ART adherence prior to PEL diagnosis, extensive mediastinal and vital organ involvement with large pleural effusion at presentation, concurrent HLH with delayed HLH treatment and disseminated MAC infection. The decision to transition to palliative care reflects the reality that, in advanced cases of PEL with significant comorbidities, curative treatment may be unattainable, and focus should shift to maintaining the patient’s quality of life.
Learning points
Extranodal extracardiac EC-PEL involving the mediastinum, especially the middle and posterior mediastinum, is rare. PEL-associated HLH is also rare, and the concurrence of these two hematologic conditions may significantly worsen patient survival odds. A high index of suspicion for EC-PEL in HIV-positive patients demonstrating a mediastinal mass, and for HLH in confirmed PEL cases, is necessary, as early diagnosis and intervention is crucial for favorable patient outcomes.
Acknowledgments
None to declare.
Financial Disclosure
None to declare.
Conflict of Interest
None to declare.
Informed Consent
Informed consent was obtained from the patient prior to death.
Author Contributions
All authors contributed to the final manuscript. CI wrote the main text under the supervision of XQ. XQ, CI and JK were involved with final pathologic diagnoses. CI, AA and AM were involved in pathologic descriptions and manuscript table and figure generation. CI, XQ and JK were involved in final manuscript edits.
Data Availability
The authors declare that data supporting the findings of this study are available within the article.
| References | ▴Top |
- Javadi T, Morales B, Olson JJ, Kothari S, Zhang L,
Abedalthagafi M. Extracavitary primary effusion lymphoma presenting as a solitary brain mass.
CNS Oncol. 2024;13(1):2357535.
doi pubmed - Carbone A, De Paoli P, Gloghini A, Vaccher E.
KSHV-associated multicentric Castleman disease: A tangle of different entities requiring
multitarget treatment strategies. Int J Cancer. 2015;137(2):251-261.
doi pubmed - Carbone A, Cesarman E, Spina M, Gloghini A, Schulz TF.
HIV-associated lymphomas and gamma-herpesviruses. Blood. 2009;113(6):1213-1224.
doi pubmed - Liao G, Cai J, Yue C, Qing X. Extracavitary/solid variant of
primary effusion lymphoma presenting as a gastric mass. Exp Mol Pathol.
2015;99(3):445-448.
doi pubmed - Cesarman E, Chang Y, Moore PS, Said JW, Knowles DM. Kaposi's
sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas.
N Engl J Med. 1995;332(18):1186-1191.
doi pubmed - Chadburn A, Hyjek E, Mathew S, Cesarman E, Said J, Knowles
DM. KSHV-positive solid lymphomas represent an extra-cavitary variant of primary effusion
lymphoma. Am J Surg Pathol. 2004;28(11):1401-1416.
doi pubmed - Katz H, Rose C, Rivera NT, Bray N. HIV-associated primary
effusion lymphoma presenting as a paracardial mass. BMJ Case Rep.
2015;2015:bcr2014208718.
doi pubmed - Zanelli M, Sanguedolce F, Zizzo M, Palicelli A, Bassi MC,
Santandrea G, Martino G, et al. Primary effusion lymphoma occurring in the setting of
transplanted patients: a systematic review of a rare, life-threatening post-transplantation
occurrence. BMC Cancer. 2021;21(1):468.
doi pubmed - Hashmi H, Murray D, Al-Quran S, Tse W. Primary effusion
lymphoma without an effusion: a rare case of solid extracavitary variant of primary effusion
lymphoma in an HIV-positive patient. Case Rep Hematol. 2018;2018:9368451.
doi pubmed - Lopez de Caceres CVB, Sant'Ana MSP, Roman Tager EMJ, Burbano
RMR, de Almeida OP, Vargas PA, Fonseca FP. Extracavitary primary effusion lymphoma affecting the
oral cavity: a rare case report. Int J Surg Pathol. 2024;32(1):119-132.
doi pubmed - Pielasinski U, Santonja C, Rodriguez-Pinilla SM, Requena L.
Extracavitary primary effusion lymphoma presenting as a cutaneous tumor: a case report and
literature review. J Cutan Pathol. 2014;41(9):745-753.
doi pubmed - Foster WR, Bischin A, Dorer R, Aboulafia DM. Human
herpesvirus type 8-associated large B-cell lymphoma: a nonserous extracavitary variant of
primary effusion lymphoma in an HIV-infected man: a case report and review of the literature.
Clin Lymphoma Myeloma Leuk. 2016;16(6):311-321.
doi pubmed - Carbone A, Gloghini A, Vaccher E, Cerri M, Gaidano G,
Dalla-Favera R, Tirelli U. Kaposi's sarcoma-associated herpesvirus/human herpesvirus type
8-positive solid lymphomas: a tissue-based variant of primary effusion lymphoma. J Mol
Diagn. 2005;7(1):17-27.
doi pubmed - Gathers DA, Galloway E, Kelemen K, Rosenthal A, Gibson SE,
Munoz J. Primary effusion lymphoma: a clinicopathologic perspective. Cancers (Basel).
2022;14(3):722.
doi pubmed - Lurain K, Polizzotto MN, Aleman K, Bhutani M, Wyvill KM,
Goncalves PH, Ramaswami R, et al. Viral, immunologic, and clinical features of primary effusion
lymphoma. Blood. 2019;133(16):1753-1761.
doi pubmed - Kastnerova L, Belousova IE, Michal M, Ptakova N, Michal M,
Kazakov DV. Kaposi sarcoma in association with an extracavitary primary effusion lymphoma
showing unusual intravascular involvement: report of a case harboring a FAM175A germline
mutation. Am J Dermatopathol. 2020;42(1):55-60.
doi pubmed - Hu Z, Pan Z, Chen W, Shi Y, Wang W, Yuan J, Wang E, et al.
Primary effusion lymphoma: a clinicopathological study of 70 cases. Cancers (Basel).
2021;13(4):878.
doi pubmed - Pan ZG, Zhang QY, Lu ZB, Quinto T, Rozenvald IB, Liu LT,
Wilson D, et al. Extracavitary KSHV-associated large B-Cell lymphoma: a distinct entity or a
subtype of primary effusion lymphoma? Study of 9 cases and review of an additional 43 cases.
Am J Surg Pathol. 2012;36(8):1129-1140.
doi pubmed - Pastore RD, Chadburn A, Kripas C, Schattner EJ. Novel
association of haemophagocytic syndrome with Kaposi's sarcoma-associated herpesvirus-related
primary effusion lymphoma. Br J Haematol. 2000;111(4):1112-1115.
doi pubmed - Epperla N, Harrington AM, Hemauer K, Shah NN. Extracavitary
primary effusion lymphoma associated with hemophagocytic lymphohistiocytosis.
Am J Hematol. 2016;91(11):1161-1164.
doi pubmed - Shah NN, Harrison N, Stonecypher M, Frank D, Amorosa V,
Svoboda J. Extracavitary primary effusion lymphoma initially presenting with hemophagocytic
lymphohistocytosis. Clin Lymphoma Myeloma Leuk. 2014;14(5):e157-160.
doi pubmed - Lee JC, Logan AC. Diagnosis and management of adult
malignancy-associated hemophagocytic lymphohistiocytosis. Cancers (Basel).
2023;15(6):1839.
doi pubmed - Lofstedt A, Jadersten M, Meeths M, Henter JI.
Malignancy-associated hemophagocytic lymphohistiocytosis in Sweden: incidence, clinical
characteristics, and survival. Blood. 2024;143(3):233-242.
doi pubmed - Carvelli J, Piperoglou C, Farnarier C, Vely F, Mazodier K,
Audonnet S, Nitschke P, et al. Functional and genetic testing in adults with HLH reveals an
inflammatory profile rather than a cytotoxicity defect. Blood. 2020;136(5):542-552.
doi pubmed - Setiadi A, Zoref-Lorenz A, Lee CY, Jordan MB, Chen LYC.
Malignancy-associated haemophagocytic lymphohistiocytosis. Lancet Haematol.
2022;9(3):e217-e227.
doi pubmed - Knauft J, Schenk T, Ernst T, Schnetzke U, Hochhaus A, La
Rosee P, Birndt S. Lymphoma-associated hemophagocytic lymphohistiocytosis (LA-HLH): a scoping
review unveils clinical and diagnostic patterns of a lymphoma subgroup with poor prognosis.
Leukemia. 2024;38(2):235-249.
doi pubmed - Aguilar C, Laberiano C, Beltran B, Diaz C, Taype-Rondan A,
Castillo JJ. Clinicopathologic characteristics and survival of patients with primary effusion
lymphoma. Leuk Lymphoma. 2020;61(9):2093-2102.
doi pubmed - Calabro ML, Sarid R. Human herpesvirus 8 and
lymphoproliferative disorders. Mediterr J Hematol Infect Dis.
2018;10(1):e2018061.
doi pubmed - Guillet S, Gerard L, Meignin V, Agbalika F, Cuccini W, Denis
B, Katlama C, et al. Classic and extracavitary primary effusion lymphoma in 51 HIV-infected
patients from a single institution. Am J Hematol. 2016;91(2):233-237.
doi pubmed
This
article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0
International License, which permits unrestricted non-commercial use, distribution, and
reproduction in any medium, provided the original work is properly cited.
Journal
of Medical Cases is published by Elmer Press Inc.