


Histoplasmosis-Associated Hemophagocytic Lymphohistiocytosis in the Setting of Granulomatosis With Polyangiitis
DOI:
https://doi.org/10.14740/jmc5126Keywords:
Secondary hemophagocytic lymphohistiocytosis, Disseminated histoplasmosis, Granulomatosis with polyangiitis, Immunosuppressive therapyAbstract
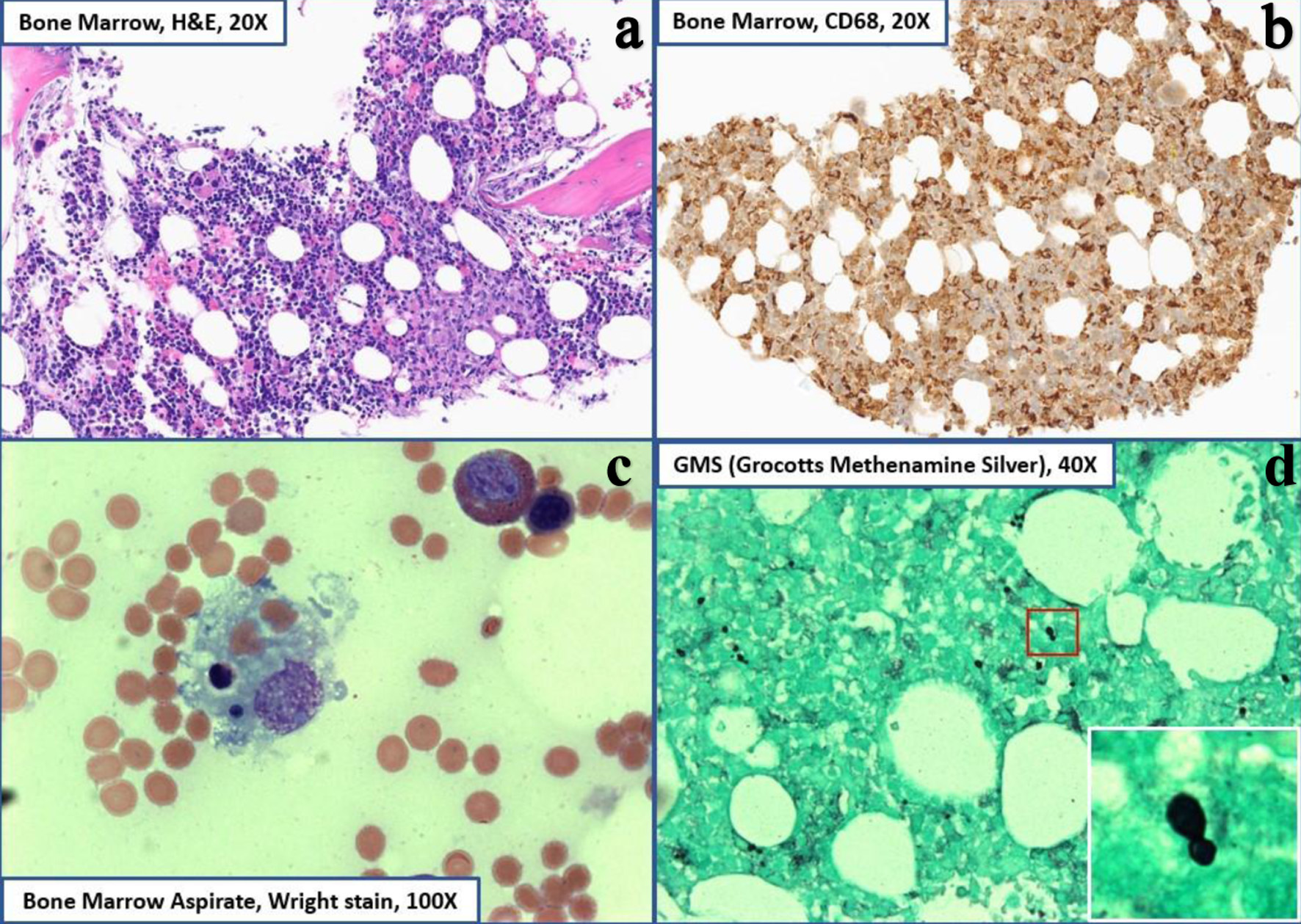
Hemophagocytic lymphohistiocytosis (HLH) is an aggressive and potentially life-threatening syndrome of aberrant immune hyperactivation characterized by excess cytokine release due to abnormal cytotoxic T-cell and macrophage activation. Secondary, or acquired, HLH occurs in the setting of underlying infection, malignancy, or autoimmune processes and often presents with systemic symptoms and multi-organ dysfunction that can initially be misattributed to infection, leading to a delay in diagnosis and management. Histoplasmosis-associated HLH is an infrequently described manifestation of secondary HLH that can occur in the setting of immunocompromised states. A 67-year-old woman with a history of granulomatosis with polyangiitis on active mycophenolate mofetil treatment initially presented with persistent flu-like symptoms in the setting of pancytopenia and elevated liver enzymes. Despite appropriate sepsis evaluation and extensive antimicrobial treatment, she remained persistently febrile and developed acute respiratory failure. Lab work revealed severely abnormal coagulation factors, hypofibrinogenemia, hyperferritinemia, and hypertriglyceridemia concerning for underlying hematologic disease process. Bone marrow biopsy obtained showed a hypercellular marrow with histiocytosis and hemophagocytic forms as well as intracellular narrow-based budding yeast, confirming an HLH diagnosis and suggestive of disseminated histoplasmosis. She was subsequently started on a high-dose steroid taper, intravenous immunoglobulin, and anti-fungal therapy with clinical improvement and stabilization of blood counts. Post-discharge follow-up has not involved repeat hospitalizations. Diagnosing HLH requires consideration of both clinical and laboratory criteria but there remains a lack of consensus use, especially in adult patients and those with associated autoimmune disease. Furthermore, the clinical presentation of HLH can overlap significantly with sepsis or other acute illnesses, thus delaying timely diagnosis and interventions for optimal patient outcomes. Our case emphasizes the importance of early consideration and comprehensive evaluation in this setting for HLH and secondary causes, including fungal etiologies. Additional clinical reporting of this rare presentation can increase clinician recognition and highlight potential diagnostic and treatment approaches. Subsequent close clinical observation with serial examinations and biochemical marker follow-up are needed to assess response and evaluate for changes in treatment approach.
Published
Issue
Section
License
Copyright (c) 2025 The authors

This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License.






