| Journal of Medical Cases, ISSN 1923-4155 print, 1923-4163 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Med Cases and Elmer Press Inc |
| Journal website https://jmc.elmerpub.com |
Case Report
Volume 17, Number 7, July 2026, pages 336-339

A Rare Genetic Association of an NPHP2 Mutation With Nephronophthisis in a Child With Bombay Blood Group
Bassil Leghrouza, b, Tariq Hindia, Ihab Hiloa, Alex Ballouta, Musa Hindiyeha
aDepartment of Pediatrics, Augusta Victoria Hospital, Jerusalem, Palestine
bCorresponding Author: Bassil Leghrouz, Department of Pediatrics, Augusta Victoria Hospital, Jerusalem, Palestine
Manuscript submitted February 14, 2026, accepted April 21, 2026, published online June 3, 2026
Short title: NPHP and Bombay Blood Group
doi: https://doi.org/10.14740/jmc5309
| Abstract | ▴Top |
Nephronophthisis is a rare genetic disorder affecting the kidneys and is one of the leading causes of end-stage kidney disease in children. The Bombay blood group is an extremely rare blood type that poses serious clinical challenges, particularly when managing patients with chronic kidney diseases. This report described a 3-year-old female patient who presented with severe anemia and kidney impairment, which progressed to end-stage kidney disease. During blood grouping, she was unexpectedly identified as having the Bombay phenotype, complicating her anemia management. Genetic testing revealed two novel pathogenic variants: one in the NPHP2 gene, associated with nephronophthisis, and another in the FUT1 gene, which is responsible for the Bombay blood group. The child received treatment involving fluid restriction and diuretic infusions. However, she subsequently progressed to end-stage kidney disease with significant fluid overload and required peritoneal dialysis. Anemia management was achieved through intravenous iron and erythropoiesis-stimulating agents (erythropoietin), eliminating the need for blood transfusions. This case underscores the rare coexistence of nephronophthisis and the Bombay blood group alongside novel mutations, highlighting the critical need for early genetic diagnosis and intervention. It also emphasizes the complexities of managing patients with such rare blood types, which complicate potential transfusion options.
Keywords: NPHP2; Nephronophthisis; Bombay blood group; Pediatric nephrology; Rare disease
| Introduction | ▴Top |
Nephronophthisis (NPHP) is a genetic kidney disorder that leads to progressive kidney disease. It is usually inherited in an autosomal recessive manner and is often seen in families with consanguineous marriages. There are several subtypes, each linked to specific genetic mutations. More than 25 genes, most of which code for proteins needed for primary cilia in kidney cells, are involved in NPHP. The main problem is tubular dysfunction, which causes symptoms like polyuria and polydipsia. It can also lead to severe anemia, poor growth, tubulointerstitial nephritis, cystic kidney disease, and eventually end-stage kidney disease (ESKD), requiring renal replacement therapy [1].
The Bombay blood group (Oh), first identified in India, is an extremely rare blood type characterized by the absence of the H antigen. Individuals with this blood group possess anti-H antibodies, rendering them unable to receive blood from any other blood group (A, B, AB, or O), which poses significant transfusion challenges.
The aim of this report is to describe the coexistence of NPHP, and the Bombay phenotype, and to highlight the clinical implications for transfusion planning and long-term management.
| Case Report | ▴Top |
A 3-year-old girl presented to the pediatric emergency department with fever, vomiting, and diarrhea. She was pale, and her complete blood count (CBC) showed a hemoglobin level of 6.6 g/dL. There was no history of hemoptysis, melena, or gross hematuria. The patient was admitted to the hospital for a workup of anemia. The repeated CBC showed a hemoglobin level of 5.8 g/dL, a white blood cell count of 9.8 × 103/mm3, and a platelet count of 301 × 103/µL. Blood work on admission is shown in Table 1.
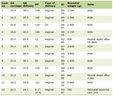 Click to view | Table 1. Blood Work on Admission |
When the patient was admitted, she had low urine output, generalized edema, and high blood pressure. Clinical and laboratory results showed signs of chronic kidney disease (CKD). Her kidney function was impaired, and she had metabolic acidosis, hyperkalemia, high parathyroid hormone, and severe anemia, even though her iron levels were normal. An ultrasound showed that both kidneys were small, with the right kidney measuring 5.6 cm and the left 6.5 cm in bipolar length. Both kidneys appeared echogenic with loss of corticomedullary differentiation.
To treat her high potassium and acidosis, she received intravenous calcium gluconate, oral Kayexalate, and intravenous sodium bicarbonate.
The patient’s parents are first cousins. They are both healthy, and there is no family history of kidney or hematological diseases.
NPHP was considered likely because it is common in the area, and the parents are related. A whole-exome sequencing (WES) test was ordered.
As part of anemia management, blood grouping identified the Bombay blood group. Due to the absence of a Bombay blood type donor, anemia was managed with an intravenous erythropoiesis-stimulating agent (ESA) and intravenous iron. The patient’s serum hemoglobin gradually improved, reaching 15 mg/dL after 3 months of treatment. During her illness course, her kidney function test continued to deteriorate, and the urine output became very minimal with significant fluid overload despite the use of diuretics. We decided to insert a peritoneal dialysis catheter and initiate dialysis, which successfully managed her fluid overload. She was discharged on home intermittent peritoneal dialysis (IPD) with regular follow-up.
Genetic testing result
WES revealed a homozygous pathogenic variant of the INVS (NPHP2) gene. A genetic diagnosis of autosomal recessive NPHP type 2 was confirmed (the mutated gene: INVS (NPHP2); variant: NM_014425.3:c.2029del, amino acid change: p.(Ser677ProfsTer66)).
The INVS variant c.2029del p.(Ser677ProfsTer66) is a frameshift variant located in exon 13 (of 17) that is predicted to create a premature stop codon and, therefore, produce a truncated protein. To the best of our knowledge, this is a novel variant that has not been previously reported in the literature. It was classified as pathogenic based on the CENTOGENE implementation of the ACMG/AMP/ClinGen SVI guidelines. Pathogenic variants of the INVS gene cause autosomal recessive NPHP type 2. (OMIM®: 602088).
WES also showed a novel homozygous missense variant in FUT1 35C>A; p.Ala12Asp, reported as a variant of uncertain significance (VUS). This mutation was further confirmed by Sanger sequencing. Sanger sequencing testing revealed that the father, mother, and the proband’s sister were all heterozygous (C/A) carriers of the same FUT1 mutation. On the paternal side, the grandmother was wild type, whereas the grandfather was a heterozygous carrier. On the maternal side, the grandmother was wild type, whereas the grandfather refused genetic testing. This inheritance pattern across three generations supports autosomal recessive transmission, consistent with the proband’s homozygous genotype and confirmed Bombay phenotype.
| Discussion | ▴Top |
This case report describes a 3-year-old girl with chronic kidney disease and severe anemia. Upon investigation, she was found to have the rare Bombay blood group caused by a mutation in the FUT1 gene, alongside another mutation in the NPHP2 gene, which is linked to a condition known as infantile NPHP.
This complex combination creates significant challenges for her treatment, particularly as she may need blood transfusions, with her unique blood type requiring careful management to avoid serious reactions. This case underscores the intricate nature of pediatric nephrology, especially in regions where genetic factors, often arising from consanguinity, lead to the prevalence of rare blood types and hereditary kidney diseases. Understanding the genetic factors involved not only clarifies the girl’s clinical situation but also highlights the critical role of genetic testing in diagnosing familial kidney disorders.
NPHP is a common genetic cause of ESKD in children, with its incidence varying widely across populations; for example, the estimated incidence varies from 1:50,000 live births in Finland to 1:1,000,000 in the United States [2]. This genetically heterogeneous disorder is associated with mutations in more than 25 identified genes [3, 4]. In approximately 10–20% of cases, patients exhibit additional features including retinal defects, liver fibrosis, skeletal abnormalities, and neurodevelopmental disorders [1].
Mutations in the NPHP1 gene are the most common, reported in approximately 20% of cases [5]. Clinically, three clinical subtypes of NPHP have been recognized based on the median age of onset of ESKD: infantile, juvenile, and adolescent/adult [6]. Infantile NPHP or NPHP type 2 (mutation in the NPHP2 gene) is one of the most significant. It can present in utero with an oligohydramnios sequence (limb contractures, pulmonary hypoplasia, and facial dysmorphisms) or postnatally with severe hypertension and kidney manifestations that progress to ESKD before the age of 3 years. It is characterized by rapid disease progression. Kidney ultrasound findings show moderately enlarged cystic kidneys with cortical hyperechogenicity [7].
The diagnosis of NPHP is suggested by clinical features and confirmed by a positive genetic test. A single-gene mutation can be detected in up to 60% of cases, depending on the cohort composition. However, the absence of a mutation is insufficient to exclude the diagnosis of NPHP. Patients with NPHP typically have a “bland” urinalysis, without evidence of proteinuria, hematuria, or cellular elements, until the late stage, when proteinuria may develop and lead to secondary glomerulosclerosis. To date, treatment options for NPHP remain limited. Blood pressure control is a priority. Management of complications arising from progressive kidney disease, such as anemia, uremia symptoms, and fluid overload, is important alongside preparation for future kidney replacement therapy.
This disease does not recur in a transplant, and kidney transplantation remains the ideal mode of kidney replacement therapy. Managing ESKD in a patient with the Bombay blood group is challenging, especially if the underlying cause of ESKD is NPHP, as these patients are commonly anemic and occasionally require blood transfusions; having the Bombay blood group prevents them from receiving blood from other blood groups. In this case, the patient responded favorably to conservative hematologic management with erythropoietin injections, thereby eliminating the risks of blood transfusion. However, comprehensive long-term planning, including the establishment of a registry for rare blood donors, remains an essential component of care for individuals with rare blood types. One option to help overcome this dilemma is to improve the patient’s hemoglobin to a good level, where his own blood can be stored for any further needs. This involves cooling blood products to very low temperatures (deep-freeze), typically between –80 °C and –96 °C. This process uses cryoprotectants that prevent ice crystal formation, which can damage cells and proteins. Deeply frozen blood products can be stored for several years. Kidney transplantation is another challenge for patients with the Bombay blood group. This difficulty arises because receiving a kidney from a donor with a different blood group carries a high risk of hemolysis and acute kidney rejection. For instance, there is a reported case from India of a patient with the Bombay blood group who successfully received a kidney from a donor with a different blood group after careful, complex strategies. These include exceptionally rare and complex meticulous planning and a precise desensitization protocol with rituximab, plasma exchange, and intravenous immunoglobulin prior to transplantation to prevent hemolytic anemia and rejection of the transplanted kidney [8].
Conclusions
This case report is the first documentation of a dual diagnosis of NPHP and Bombay blood group. In addition, the two mutations reported in this case are novel. This case highlights the need for specialized clinical management that accommodates genetic considerations and the unique immunological profile of the patient, as the child responded favorably to intravenous iron and ESA. This report aims to discuss the therapeutic implications and interdisciplinary approach to managing such complex cases.
Acknowledgments
We thank the patient and her family for their cooperation and consent to publish this report. We acknowledge the support of the Pediatric Nephrology, Pediatric Hematology, and Genetic Laboratory staff for their assistance in patient diagnosis and management of the patient.
Financial Disclosure
No external funding or financial support was received for this manuscript.
Conflict of Interest
The authors declared that the research was conducted in the absence of any commercial or financial relationships that could be a potential conflict of interest.
Informed Consent
The authors confirmed that written informed consent for publication was obtained from the patient’s legal guardian. The study was approved by the Institutional Review Board (IRB).
Author Contributions
Bassil Leghrouz: corresponding author, case review, literature review, manuscript writing. Tareq Hindi: manuscript review, review of NPHP2 literature. Ihab Hilo: writing of the genetic section, manuscript review. Alex Ballout: review of Bombay blood group literature, writing of the genetic section, manuscript review. Musa Hindiyeh: review of Bombay blood group literature, writing of the genetic section, manuscript review.
Data Availability
The data supporting the findings of this study are included within the article. Additional details are available from the corresponding author upon reasonable request, in accordance with patient confidentiality and ethical restrictions.
Abbreviations
NPHP: nephronophthisis; ESKD: end-stage kidney disease; CKD: chronic kidney disease; WES: whole-exome sequencing; ESA: erythropoiesis-stimulating agent; IPD: intermittent peritoneal dialysis; VUS: variant of uncertain significance
| References | ▴Top |
- Luo F, Tao YH. Nephronophthisis: A review of genotype-phenotype correlation. Nephrology (Carlton). 2018;23(10):904-911.
doi pubmed - Ala-Mello S, Koskimies O, Rapola J, Kaariainen H. Nephronophthisis in Finland: epidemiology and comparison of genetically classified subgroups. Eur J Hum Genet. 1999;7(2):205-211.
doi pubmed - Macia MS, Halbritter J, Delous M, Bredrup C, Gutter A, Filhol E, Mellgren AEC, et al. Mutations in MAPKBP1 Cause Juvenile or Late-Onset Cilia-Independent Nephronophthisis. Am J Hum Genet. 2017;100(2):323-333.
doi pubmed - Tory K, Lacoste T, Burglen L, Moriniere V, Boddaert N, Macher MA, Llanas B, et al. High NPHP1 and NPHP6 mutation rate in patients with Joubert syndrome and nephronophthisis: potential epistatic effect of NPHP6 and AHI1 mutations in patients with NPHP1 mutations. J Am Soc Nephrol. 2007;18(5):1566-1575.
doi pubmed - Halbritter J, Bizet AA, Schmidts M, Porath JD, Braun DA, Gee HY, McInerney-Leo AM, et al. Defects in the IFT-B component IFT172 cause Jeune and Mainzer-Saldino syndromes in humans. Am J Hum Genet. 2013;93(5):915-925.
doi pubmed - Hildebrandt F, Attanasio M, Otto E. Nephronophthisis: disease mechanisms of a ciliopathy. J Am Soc Nephrol. 2009;20(1):23-35.
doi pubmed - Stokman M, Lilien M, Knoers N. Nephronophthisis-Related Ciliopathies. In: Adam MP, Bick S, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, eds. GeneReviews((R)). Seattle (WA). 1993.
pubmed - Raj TY, Kanakaraj A, Jeyakumar JD, Manikandan R, Subash C, Ravichandran R. Kidney transplantation across ABO-H incompatibility in a recipient with Bombay blood group: A novel report. Am J Transplant. 2026;26(3):646-649.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (CC BY 4.0), which permits unrestricted use, distribution, and reproduction in any medium, including commercial use, provided the original work is properly cited.
Journal of Medical Cases is published by Elmer Press Inc.