| Journal of Medical Cases, ISSN 1923-4155 print, 1923-4163 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Med Cases and Elmer Press Inc |
| Journal website https://jmc.elmerpub.com |
Case Report
Volume 17, Number 6, June 2026, pages 263-270

Isolated Neutropenia as a Sentinel of High-Risk Clonal Evolution: Acute Myeloid Leukemia With Myelodysplasia-Related Changes Harboring TP53 Deletion via Isochromosome 17q and Deletion 20q Mimicking a Myeloproliferative Neoplasm
Caryn Louise D. Gutierreza, b, Anne Kristine H. Quero-Taggaoaa
aDivision of Hematology, Department of Internal Medicine, Philippine General Hospital, Manila, Philippines
bCorresponding Author: Caryn Louise D. Gutierrez, Division of Hematology, Department of Internal Medicine, Philippine General Hospital, Manila, Philippines
Manuscript submitted January 31, 2026, accepted March 16, 2026, published online April 29, 2026
Short title: Isolated Neutropenia Signaling High-Risk AML
doi: https://doi.org/10.14740/jmc5297
| Abstract | ▴Top |
Isolated neutropenia is often deemed benign in elderly patients, frequently attributed to age-related marrow changes, medications, or nutritional deficiencies. However, persistent and unexplained neutropenia may signal early clonal hematopoiesis or evolving myeloid malignancy. While acute myeloid leukemia with myelodysplasia-related changes (AML-MRC) usually arises from multilineage cytopenia, presentation with isolated neutropenia and myeloproliferative neoplasia (MPN)-like features is exceedingly rare. This case underscores the importance of early, risk-adapted assessment in such scenarios. A 69-year-old woman presented with isolated mild neutropenia (absolute neutrophil count (ANC) 1,188/µL) and easy bruising. With no other cytopenias or symptoms, bone marrow biopsy was deferred. Six months later, she developed abdominal discomfort, anorexia, and massive splenomegaly. Laboratory tests revealed leukocytosis with left shift, anemia, thrombocytopenia, and nucleated red cells. Bone marrow biopsy showed > 40% myeloblasts, trilineage dysplasia, and mild fibrosis. JAK2 mutation and BCR-ABL were negative. Cytogenetics demonstrated i(17q) and del(20q); Fluorescence in situ hybridization (FISH) showed TP53 deletion in 81.91% of nuclei, establishing AML-MRC with high-risk features. She was started on azacitidine and venetoclax. Day 28 marrow showed minimal residual disease (blast 8%) but no metaphase growth prompting treatment continuation. She is currently on her fourth cycle with treatment-related transfusion-dependent cytopenias, prompting dose reduction. This case challenges the prevailing “watch-and-wait” paradigm in the evaluation of isolated neutropenia, particularly in older adults. It exposes the hidden risk of clinical inertia, where the absence of other overt cytopenias or symptoms may lead to missed opportunities for early diagnosis of high-risk clonal evolution. In resource-limited setting, this case underscores urgent need for heightened clinical vigilance. Likewise, it highlights the urgent need for a more risk-adapted approach to persistent isolated neutropenia and compels us to confront a critical question: how many patients are we failing to diagnose in time, and how many windows for potentially life-altering intervention are we silently allowing to close?
Keywords: Isolated neutropenia; Clonal evolution; Acute myeloid leukemia with myelodysplasia related changes; TP53 deletion; Isochromosome 17q; Deletion 20q; Leukoerythroblastosis; High-risk cytogenetics
| Introduction | ▴Top |
Isolated neutropenia is frequently overlooked in clinical practice, particularly among elderly patients, where it is often attributed to benign causes such as medications, infections, nutritional deficiencies (e.g., folate and vitamin B12), immune-mediated conditions, or age-related causes [1, 2]. In the absence of additional cytopenias or overt morphologic dysplasia, this subtle manifestation often leads to a delayed or deferred diagnostic workup [3, 4]. In such contexts, especially in a resource-constrained setting, stable patients with persistent isolated neutropenia are frequently managed conservatively, with a “watch and wait” approach grounded in the assumption that the risk of malignant evolution is low [5]. Persistent isolated neutropenia without an identifiable cause may represent the earliest clinical footprint of a clonal myeloid disorder. When persistent, severe and unexplained, isolated neutropenia, although uncommon, may serve as an early harbinger of clonal hematopoiesis or a pre-malignant myeloid condition such as myelodysplastic syndrome (MDS) or clonal cytopenia of undetermined significance (CCUS) [2, 3, 6]. Isolated neutropenia in MDS is rare occurring in only about 2% of cases [7]. Even when these clonal precursors are identified, the current paradigm continues to favor observation over intervention [6, 7].
Acute myeloid leukemia with myelodysplasia-related changes (AML-MRC) is one of the more commonly encountered subtypes of AML, accounting for approximately 20% of all cases [8]. These patients generally have poorer prognosis and demonstrate lower response rates to therapy particularly when associated with high-risk cytogenetic abnormalities such as TP53 deletion [8, 9]. AML-MRC is defined by > 20% bone marrow blasts plus either a history of MDS/myeloproliferative neoplasia (MPN), MDS-related cytogenetics, or ≥ 50% multilineage dysplasia. Prior chemotherapy, radiation, or recurrent AML-defining cytogenetics exclude the diagnosis [8, 10]. In general, AML-MRC evolves from an antecedent hematologic condition, or overt cytopenias [10, 11]. Considering these points, it raises a crucial yet unresolved question in our current hematologic practice: should we intervene early in cases of unexplained neutropenia, or continue to observe under the prevailing “watch and wait” strategy?
We present a rare case that challenges this paradigm: a case of AML-MRC in an elderly female who initially presented with subtle isolated neutropenia and was closely observed, but subsequently developed aggressive AML with massive splenomegaly, leukoerythroblastosis, and mild marrow fibrosis—features more typical of primary myelofibrosis (PMF) or MDS/MPN overlap syndrome. Cytogenetic analysis revealed an extremely rare coexistence of i(17q) and del(20q), supporting the presence of high-risk clonal evolution. To our knowledge, this is the first documented case of isolated neutropenia progressing to AML-MRC with MPN-like features. This underscores the importance of clinical vigilance and re-evaluation of current thresholds for invasive diagnostics in otherwise stable patients. It also raises important questions about whether early therapeutic intervention might have altered the trajectory of disease progression in cases of isolated neutropenia—a consideration particularly relevant in settings where delayed diagnosis is often the rule rather than the exception.
| Case Report | ▴Top |
A 69-year-old female initially presented with occasional episodes of easy bruising in the absence of trauma. Her initial complete blood count (CBC) was notable only for mild neutropenia, with a hemoglobin of 125 g/L, hematocrit 42%, white blood cell (WBC) count of 3.6 × 109/L, absolute neutrophil count (ANC) of 1,188/µL, and platelet count of 217 × 109/L. Coagulation studies were within normal limits. She was diagnosed with senile purpura, with an International Society on Thrombosis and Hemostasis–Bleeding Assessment Tool (ISTH-BAT) score of 2. She was advised to undergo routine monitoring in the outpatient department every 3 months.
At her second follow-up, the patient denied recurrence of easy bruising; however, repeat CBC showed persistent but slight worsening of neutropenia: WBC 2.5 × 109/L, ANC 803/µL, hemoglobin 129 g/L, hematocrit 43%, and a platelet count of 437 × 109/L. Peripheral blood smear (PBS) was performed, revealing a normocytic, normochromic red blood cells (RBCs), and there was a note of presence of occasional polymorphonuclear cell (PMNs) with hyposegmentation and hypogranulation. MDS was considered, and she was advised to undergo bone marrow aspiration (BMA) and biopsy. However, given the absence of transfusion requirements, additional cytopenias, or indications for an immediate change in therapeutic management, the patient declined the biopsy at that time and chose to continue with periodic laboratory monitoring instead. She was advised to do CBC monitoring every 2 months.
At her third follow-up visit, CBC showed slightly improved leukocyte counts but persistent mild neutropenia (WBC 3.2 × 109/L, ANC 1,088/µL), with no anemia (hemoglobin 126 g/L) or thrombocytopenia (platelet count 163 × 109/L). Although still within the normal reference range, the platelet count represented a decline from the previously elevated value of 437 × 109/L. PBS findings remained consistent with prior examination, demonstrating occasional hyposegmented and hypogranulated PMNs without other significant abnormalities. The patient reported new-onset bloating and anorexia. However, abdominal examination remained unremarkable, with no palpable hepatosplenomegaly. BMA with biopsy was offered, but she did not provide consent for the procedure. Peripheral blood MDS fluorescence in situ hybridization (FISH) testing was subsequently proposed. At that time, the patient was unable to undergo the test due to financial constraints but indicated willingness to proceed once resources became available.
One month later, the patient presented to the emergency department with generalized weakness and abdominal pain. On abdominal examination, the spleen was palpable 14 cm below the left subcostal margin and extending beyond midline. Repeat CBC revealed marked leukocytosis (WBC 39.7 × 109/L) with a left-shifted differential: neutrophils 41%, lymphocytes 8%, monocytes 2%, band forms 3%, metamyelocytes 1%, myelocytes 7%, promyelocytes 10%, and blasts 28%. Anemia (hemoglobin 92 g/L) and thrombocytopenia (platelets 109 × 109/L) were also noted. PBS at this time revealed normocytic, normochromic RBCs along with the presence of nucleated red blood cells (nRBCs), and some dacryocytes. There were also occasional dysplastic PMNs characterized by hyposegmentation and hypogranulation. Whole abdominal ultrasound confirmed massive splenomegaly. Figure 1 shows the WBC count, and ANC trends of the patient during subsequent follow-ups, while Figure 2 shows the hemoglobin and platelet count trends.
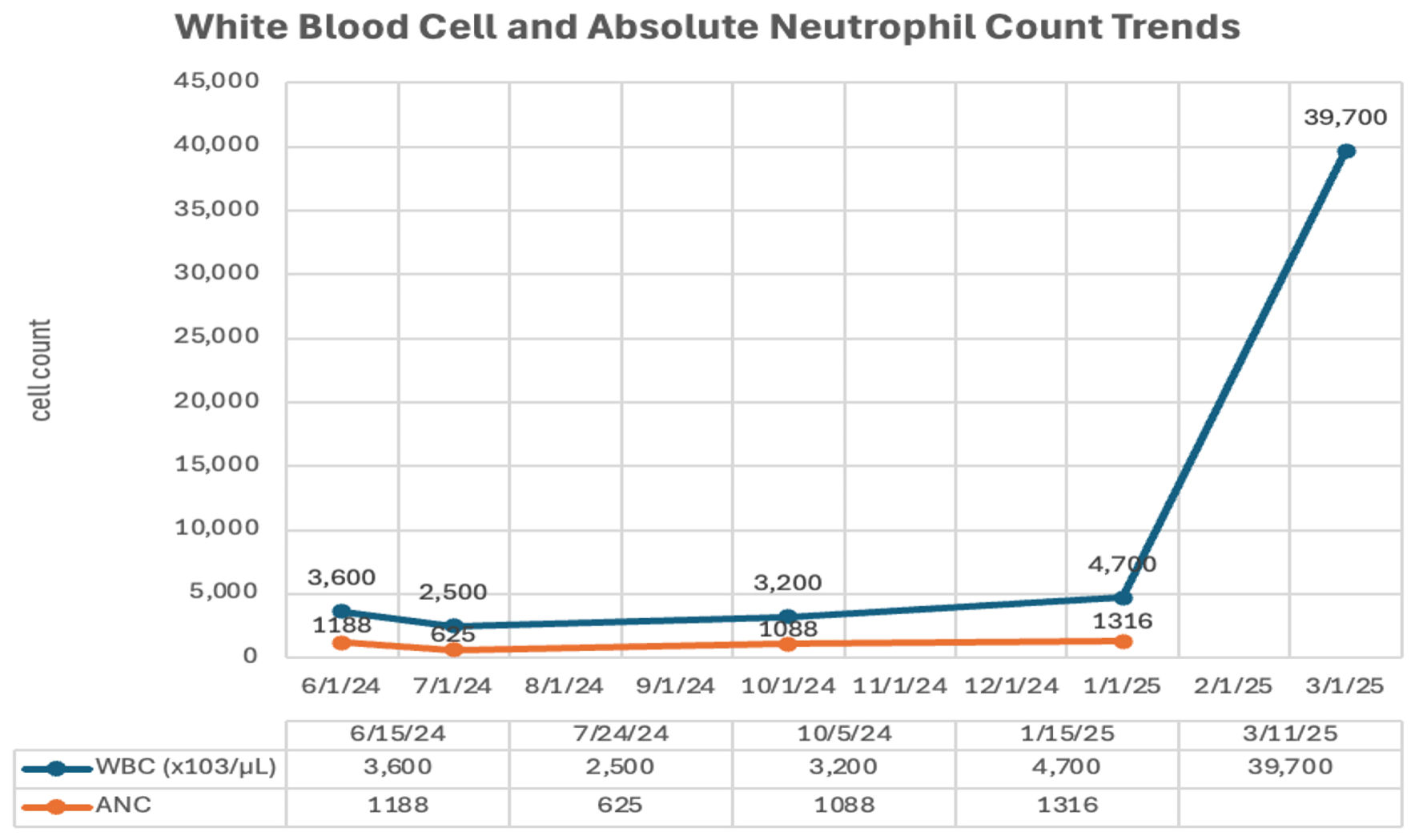 Click for large image | Figure 1. White blood cell (WBC) count and absolute neutrophil count (ANC) trends on outpatient follow-up visits. |
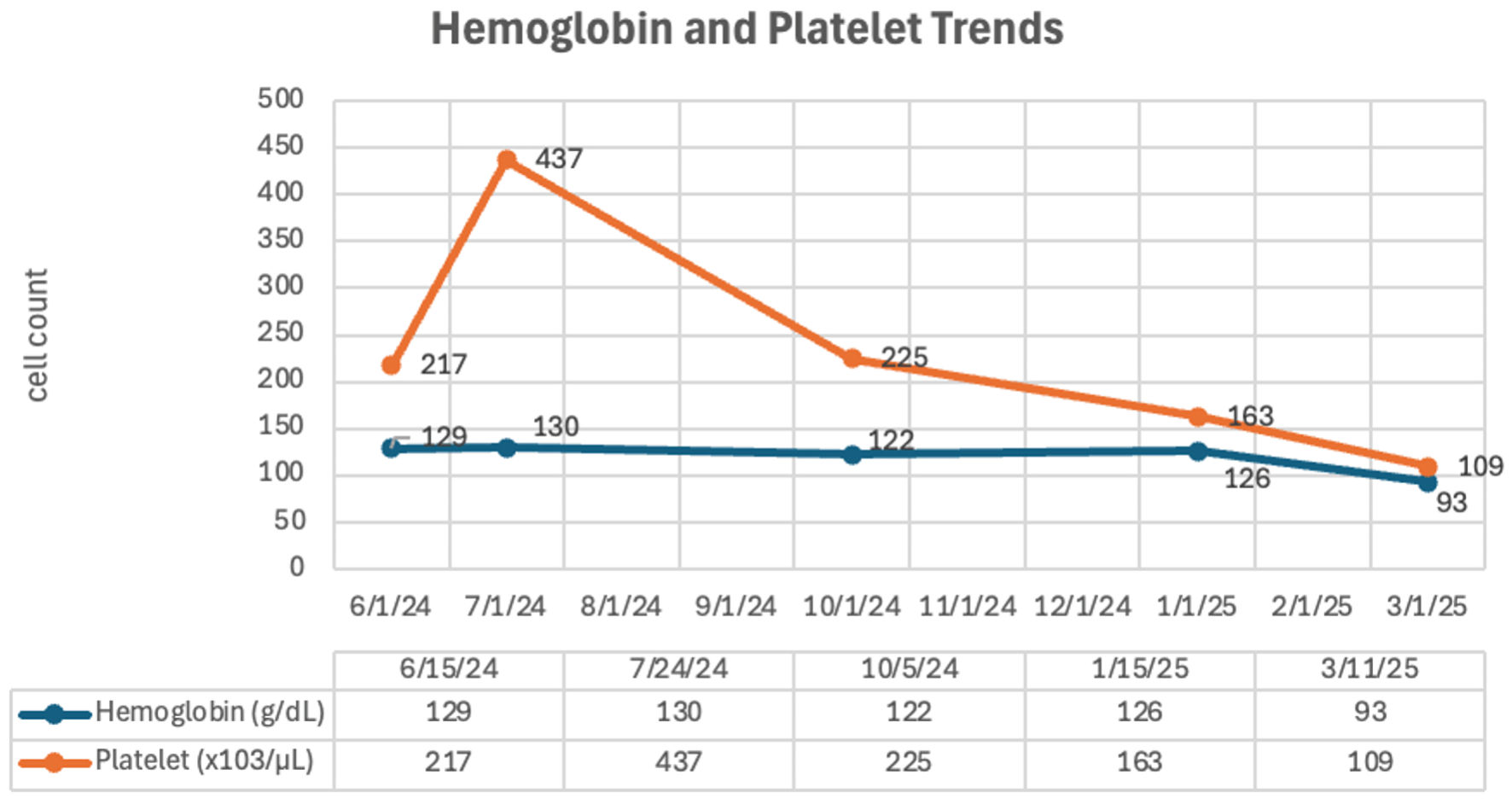 Click for large image | Figure 2. Hemoglobin and platelet trends on outpatient follow-up visits. |
Investigations
Given the combination of massive splenomegaly, and leukoerythroblastosis, MDS/MPN overlap with leukemic conversion was initially considered. However, further molecular workup, including BCR-ABL1 and JAK2 mutation testing, returned negative.
BMA and biopsy revealed a markedly hypercellular marrow for age (90% cellularity), with sheets of mononuclear cells exhibiting round to oval nuclei, fine chromatin, occasional prominent nucleoli, and high nuclear-to-cytoplasmic ratios, comprising 41–50% of marrow cellularity—consistent with myeloblasts. Trilineage dysplasia was observed, along with mild reticulin fibrosis. These findings were diagnostic of AML. Flow cytometry using a basic leukemia panel confirmed the diagnosis.
Cytogenetic analysis revealed isochromosome 17q and deletion of the long arm of chromosome 20 (del(20q)) in all 17 metaphase cells examined (Fig. 3). FISH showed a TP53 deletion in 81.91% of nuclei. FLT3 mutation was negative, and next-generation sequencing (NGS) was not performed due to financial constraints.
 Click for large image | Figure 3. Cytogenetic study showing isochromosome 17q and deletion 20q. |
Treatment
She was ultimately diagnosed with AML-MRC, harboring high-risk cytogenetic features. She was initiated on combination therapy with azacitidine and venetoclax. Day 28 bone marrow assessment following the first cycle showed minimal residual disease, and the repeat karyotyping showed no metaphase growth. This prompted continuation of treatment. However, she subsequently developed transfusion-requiring severe thrombocytopenia and severe anemia, necessitating dose modifications: azacitidine was reduced to 50% of the original dose, and venetoclax was adjusted to 100 mg every other day.
Follow-up and outcomes
At the time of writing, approximately 1 year after diagnosis, the patient was transitioned from azacitidine plus venetoclax to decitabine (10-day schedule) combined with venetoclax for 14 days due to persistent transfusion dependence. She is currently receiving her second cycle of this regimen with close monitoring for hematologic toxicity and treatment response. Early follow-up has demonstrated a notable reduction in transfusion requirements, suggesting clinical benefit from the modified therapy.
| Discussion | ▴Top |
This case highlights the clinical significance of persistent isolated neutropenia as a potential early manifestation of high-risk clonal evolution, culminating in a rare and diagnostically challenging presentation of AML-MRC with MDS/MPN-like features. It reinforces the need for a paradigm shift in how mild, unexplained neutropenia—particularly in older adults—is approached in clinical practice.
In one study by Njue et al, among patients with chronic neutropenia, the majority had idiopathic (50%) or autoimmune (33%) etiologies, while a smaller proportion had congenital forms (17%) [12]. MDS and AML rarely present as isolated neutropenia and are present only in about 2% of the cases [7, 13]. As a result, patients with isolated neutropenia are commonly monitored without extensive investigation, particularly in resource-limited settings where access to diagnostic tools is constrained. However, this conservative approach may delay recognition of clonal hematopoietic disorders, especially in cases where neutropenia is persistent and unexplained. In the present case, neutropenia remained the sole hematologic abnormality for several months, which delayed further investigation and ultimately allowed a high-risk myeloid neoplasm to progress undetected.
Emerging data underscore the importance of reassessing this approach, especially in the context of clonal hematopoiesis of indeterminate potential (CHIP) and CCUS [13]. These clonal states are more common in the elderly [14, 15]. Even in a well-resourced setting, earlier bone marrow examination and molecular profiling might have revealed the presence of TP53 mutation or other high-risk features; however, the recommendation for these diseases will still focus on closer surveillance without initiating treatment [16]. These modalities are often unavailable or cost-prohibitive in resource-constrained settings. This case powerfully demonstrates how isolated neutropenia can act as a sentinel event in clonal evolution, preceding overt leukemic transformation. The patient’s subsequent presentation with massive splenomegaly, leukoerythroblastosis, and high-risk cytogenetics (TP53 deletion via isochromosome 17q and del(20q)) exemplifies how an aggressive disease trajectory can go unrecognized in the absence of early suspicion.
In 2008, the World Health Organization (WHO) formally recognized AML-MRC as a distinct subtype within its classification system [10]. AML-MRC is generally considered biologically linked to MDS and was previously referred to as refractory anemia with excess blasts in transformation (RAEB-T) under the French-American-British (FAB) classification [17]. As shown in Table 1, the diagnosis of AML-MRC requires a bone marrow blast percentage exceeding 20% along with at least one of the following: a documented history of MDS or MPN, MDS-associated cytogenetic abnormalities, or dysplasia involving 50% or more of cells in at least two hematopoietic lineages [10, 17]. In this patient, AML-MRC was diagnosed based on the presence of > 40% blasts, multilineage dysplasia, and MDS-related cytogenetic abnormalities, specifically isochromosome 17q (i(17q)), and deletion 20q.
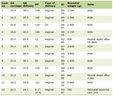 Click to view | Table 1. Diagnostic Criteria for AML-MRC |
Prognostically, AML-MRC represents one of the highest-risk AML subtypes. Multiple studies, including large-scale analyses by Takeda et al and the European LeukemiaNet (ELN), consistently show inferior complete remission (CR) rates, higher relapse risk, and shorter overall survival, particularly in patients with adverse cytogenetics such as TP53 mutation/deletion or complex karyotypes [18, 19].
The clinical presentation of this case was complicated by features more typically associated with MPNs, namely massive splenomegaly, mild marrow fibrosis, and a leukoerythroblastic blood smear. These findings are unusual in AML-MRC, which more often presents with cytopenias, dysplasia, and a hypercellular marrow without significant fibrosis or extramedullary hematopoiesis. Massive splenomegaly and leukoerythroblastosis are classical hallmarks of PMF and other chronic MPNs, reflecting extramedullary hematopoiesis and marrow exhaustion [20, 21]. The presence of reticulin fibrosis in the marrow further supported an MPN-like phenotype. However, in the absence of common MPN-associated mutations such as JAK2 V617F, and with negative BCR-ABL1, classical MPNs such as PMF, polycythemia vera (PV), essential thrombocythemia (ET), and chronic myeloid leukemia (CML) were ruled out, and the absence of monocytosis or basophilia helped exclude chronic myelomonocytic leukemia (CMML) and atypical CML (aCML) [22, 23].
The cytogenetic profile in this patient was particularly striking and highlights the aggressive biology underlying her disease. Isochromosome 17q is a rare structural chromosomal abnormality formed by duplication of the long arm and simultaneous loss of the short arm of chromosome 17, resulting in trisomy of 17q and monosomy of 17p. Importantly, the short arm of chromosome 17 (17p) harbors the TP53 tumor suppressor gene. Thus, i(17q) results in loss of TP53, either through monosomy 17p or through compound lesions with TP53 mutations or deletions. The leukemogenic potential of i(17q) is thought to stem from both the loss of tumor suppressor function (via TP53 haploinsufficiency or deletion) and the gain of dosage-sensitive genes on 17q, such as MSI2 or the granulocyte colony-stimulating factor receptor (G-CSFR), which promote myeloid proliferation [24]. In AML, i(17q) is found in approximately 0.5–1% of cases and is strongly associated with poor prognosis, refractory disease, and short overall survival, especially when combined with other cytogenetic abnormalities or TP53 disruption [25, 26]. In our patient, the presence of i(17q) in all 17 metaphases examined suggests a dominant clone, supporting its role as a primary driver of disease biology. The identification of TP53 deletion in > 80% of interphase nuclei by FISH in this patient represents a critical prognostic determinant. TP53 alterations are associated with poor response to chemotherapy (especially to anthracyclines and cytarabine), low rates of CR, high relapse rates, and median overall survival often less than 6 months, even with aggressive therapy or allogeneic stem cell transplantation [27, 28].
On the other hand, deletion of the long arm of chromosome 20 is a well-characterized cytogenetic abnormality in MDS, MPN, and less frequently in AML. It is often considered a favorable-risk lesion when isolated, but its prognostic impact changes dramatically when found in association with other abnormalities, particularly complex karyotypes [29]. The minimal commonly deleted region (MCDR) in 20q includes the L3MBTL1 gene, which acts as a chromatin modifier and tumor suppressor. Its loss has been implicated in ineffective hematopoiesis and progression to AML [30]. In this case, the coexistence of del(20q) with i(17q) suggests a multi-hit pathogenesis, where del(20q) may represent an early lesion in a pre-leukemic clone, followed by acquisition of i(17q) and TP53 deletion that drives transformation to overt AML. Interestingly, to our knowledge, there are no reported cases in the literature describing the co-occurrence of i(17q) and del(20q). The rarity of this cytogenetic profile makes this case both reportable and educational for the hematology community.
The use of hypomethylating agents (HMAs) plus venetoclax in this patient aligns with emerging data suggesting some benefit in TP53-mutated AML, though responses are often transient [31, 32]. Studies such as the VIALE-A trial demonstrated a median overall survival of just 5–7 months in TP53-mutated patients receiving azacitidine-venetoclax, highlighting the urgent need for novel strategies in this subset [33].
Beyond its cytogenetic and phenotypic complexity, this case also draws attention to the challenges of timely diagnosis—especially in resource-limited settings, where access to bone marrow biopsy, cytogenetic studies, and NGS is often constrained. In such contexts, patients presenting with persistent but stable cytopenias may not undergo adequate evaluation until transformation to overt leukemia occurs, missing a crucial window for early diagnosis or intervention. The prevailing “watchful waiting” paradigm for isolated neutropenia may thus delay recognition of CHIP or CCUS—entities now acknowledged as precursors to myeloid malignancies and frequently associated with high-risk mutations such as TP53, ASXL1, RUNX1, and SRSF2 [14, 34]. Importantly, however, even in well-resourced settings, the current standard of care for patients with CCUS remains close monitoring rather than early therapeutic intervention, due to the limited evidence that early treatment alters the natural history of disease or improves outcomes [34, 35]. Thus, despite growing molecular insight into clonal evolution, both systemic barriers in low-resource environments and the absence of established interventional guidelines in high-resource centers contribute to diagnostic inertia—underscoring the urgent need for risk-adapted strategies that can identify patients who might benefit from earlier evaluation and possibly pre-emptive therapy.
This case reinforces the need to reconsider current thresholds for invasive diagnostics in older patients with persistent neutropenia, even when other blood indices are preserved. It demonstrates that isolated neutropenia can represent not just an innocuous laboratory finding but a sentinel event in the trajectory of clonal evolution toward aggressive AML. The presence of MPN-like clinical features further illustrates the overlapping spectrum of myeloid neoplasms and underscores the critical role of integrating morphologic, cytogenetic, and molecular diagnostics in navigating these blurred boundaries.
Learning points
Persistent isolated neutropenia in older adults warrants careful evaluation, as it may represent early clonal hematopoiesis or evolving myeloid neoplasm rather than a benign finding.
Clinical inertia should be avoided when neutropenia persists without clear etiology, even in the absence of other cytopenias or symptoms.
Myeloid neoplasms may initially present with subtle hematologic abnormalities, and progression to AML with myelodysplasia-related changes can occur after a prolonged indolent phase.
Limited access to bone marrow, cytogenetic, and molecular testing in resource-constrained settings may delay diagnosis and worsen outcomes in patients with evolving clonal hematologic disease.
A risk-adapted and vigilant approach to unexplained neutropenia may facilitate earlier diagnosis and timely intervention.
Careful review of the PBS by an experienced morphologist is essential for detecting early dysplastic changes.
Acknowledgments
The authors express their gratitude to the medical and nursing teams involved in the diagnosis and management of the patient, as well as to the Department of Hematology for their valuable clinical and diagnostic support.
Financial Disclosure
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Conflict of Interest
The authors declare no conflicts of interest related to this publication.
Informed Consent
Written informed consent was obtained from the patient for publication of this case report and any accompanying images. Identifying information has been omitted to preserve confidentiality.
Author Contributions
Gutierrez, Caryn Louise: conceptualization, data collection, writing—original draft. Quero-Taggaoa, Anne Kristine: literature review, data verification, writing—review and editing. All authors have read and approved the final version of the manuscript.
Data Availability
All data supporting the findings of this case report are included within the article.
Abbreviations
aCML: atypical chronic myeloid leukemia; AML: acute myeloid leukemia; AML-MRC: acute myeloid leukemia with myelodysplasia-related changes; ANC: absolute neutrophil count; ISTH-BAT: International Society on Thrombosis and Hemostasis–Bleeding Assessment Tool; BCR-ABL1: BCR-ABL1 fusion gene (Philadelphia chromosome); CBC: complete blood count; CCUS: clonal cytopenia of undetermined significance; CHIP: clonal hematopoiesis of indeterminate potential; CML: chronic myeloid leukemia; CMML: chronic myelomonocytic leukemia; CR: complete remission; del(20q): deletion of the long arm of chromosome 20; ELN: European LeukemiaNet; ET: essential thrombocythemia; FAB: French-American-British classification; FISH: fluorescence in situ hybridization; FLT3: FMS-like tyrosine kinase 3; HMA: hypomethylating agent; i(17q): isochromosome 17q; JAK2: Janus kinase 2; MCDR: minimal commonly deleted region; MDS: myelodysplastic syndrome; MDS/MPN: myelodysplastic/myeloproliferative neoplasm; MPN: myeloproliferative neoplasm; NGS: next-generation sequencing; nRBCs: nucleated red blood cells; PMF: primary myelofibrosis; PV: polycythemia vera; RAEB-T: refractory anemia with excess blasts in transformation; TP53: tumor protein p53 gene; WBC: white blood cell; WHO: World Health Organization
| References | ▴Top |
- Keklik M, Yuce MM, Akyol G, Sanli NM, Celik S, Eser B. Causes of acquired isolated neutropenia in adulthood: a single center study. Journal of Current Hematology & Oncology Research. 2024;2(3):59-62.
doi - Steensma DP, Bejar R, Jaiswal S, Lindsley RC, Sekeres MA, Hasserjian RP, Ebert BL. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood. 2015;126(1):9-16.
doi pubmed - Sperling AS, Gibson CJ, Ebert BL. The genetics of myelodysplastic syndrome: from clonal haematopoiesis to secondary leukaemia. Nat Rev Cancer. 2017;17(1):5-19.
doi pubmed - Valent P. ICUS, IDUS, CHIP and CCUS: diagnostic criteria, separation from MDS and clinical implications. Pathobiology. 2019;86(1):30-38.
doi pubmed - Weeks LD, Ebert BL. Causes and consequences of clonal hematopoiesis. Blood. 2023;142(26):2235-2246.
doi pubmed - Fabre MA, Vassiliou GS. The lifelong natural history of clonal hematopoiesis and its links to myeloid neoplasia. Blood. 2024;143(7):573-581.
doi pubmed - Park S, Andrieu V, Sapena R, Leroux G, Stamatoullas A, Callat M, Beyne-Rauzy O, et al. MDS with isolated neutropenia or thrombocytopenia. Incidence and Characteristics in the Groupe Francophone des Myelodysplasies (GFM) registry. Blood. 2007;110(11):2456.
doi - Shibusawa M, Tanimoto T. Acute myeloid leukemia with myelodysplasia related changes. In: Li W, editor. Leukemia [Internet]. Brisbane (AU): Exon Publications; Oct 16, 2022. Chapter 6. Available from: https://www.ncbi.nlm.nih.gov/books/NBK586202/.
doi - Chow S, Misura A, Bigun O, Usta S, Sasso R, Gowlett-Park D, Czibere K, et al. Outcomes of patients with acute myeloid leukemia with myelodysplasia related changes (AML-MRC) receiving intensive Vs. non-intensive first line treatment. Blood. 2023;142(Supplement 1):5840.
doi - Kaivers J, Peters J, Rautenberg C, Schroeder T, Kobbe G, Hildebrandt B, Haas R, et al. The WHO 2016 diagnostic criteria for Acute Myeloid leukemia with myelodysplasia related changes (AML-MRC) produce a very heterogeneous entity: A retrospective analysis of the FAB subtype RAEB-T. Leuk Res. 2022;112:106757.
doi pubmed - Ogbue OD, Kewan T, Bahaj WS, Gurnari C, Visconte V, Maciejewski JP. New approaches to idiopathic neutropenia in the era of clonal hematopoiesis. Exp Hematol Oncol. 2023;12(1):42.
doi pubmed - Njue L, Porret N, Schnegg-Kaufmann AS, Varra LF, Andres M, Rovo A. Isolated severe neutropenia in adults, evaluation of underlying causes and outcomes, real-world data collected over a 5-year period in a tertiary referral hospital. Medicina (Kaunas). 2024;60(10).
doi pubmed - Newburger PE, Dale DC. Evaluation and management of patients with isolated neutropenia. Semin Hematol. 2013;50(3):198-206.
doi pubmed - Malcovati L, Galli A, Travaglino E, Ambaglio I, Rizzo E, Molteni E, Elena C, et al. Clinical significance of somatic mutation in unexplained blood cytopenia. Blood. 2017;129(25):3371-3378.
doi pubmed - Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, Lindsley RC, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med. 2014;371(26):2488-2498.
doi pubmed - Taborda CC, Zeidan AM, Mendez LM. Clonal cytopenia of undetermined significance: definitions, risk and therapeutic targets. Frontiers in Hematology. 2024;3.
doi - Bennett JM, Catovsky D, Daniel MT, Flandrin G, Galton DA, Gralnick HR, Sultan C. Proposals for the classification of the myelodysplastic syndromes. Br J Haematol. 1982;51(2):189-199.
pubmed - Dohner H, Wei AH, Appelbaum FR, Craddock C, DiNardo CD, Dombret H, Ebert BL, et al. Diagnosis and management of AML in adults: 2022 recommendations from an international expert panel on behalf of the ELN. Blood. 2022;140(12):1345-1377.
doi pubmed - Takeda J, Iwasaki M, Kanda J, Nannya Y, Hiramoto N, Kondo T, Ishikawa T, et al. Prognostic impact of chromosomal abnormalities in TP53-Mutated acute myeloid leukemia and High-Risk Myelodysplastic syndromes. Blood. 2023;142(Supplement 1):4328.
doi - Szuber N, Orazi A, Tefferi A. Chronic neutrophilic leukemia and atypical chronic myeloid leukemia: 2024 update on diagnosis, genetics, risk stratification, and management. Am J Hematol. 2024;99(7):1360-1387.
doi pubmed - Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, Bloomfield CD, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127(20):2391-2405.
doi pubmed - Vainchenker W, Kralovics R. Genetic basis and molecular pathophysiology of classical myeloproliferative neoplasms. Blood. 2017;129(6):667-679.
doi pubmed - Syeed N. JAK2 and Beyond: Mutational Study of JAK2V617 in Myeloproliferative Disorders and Haematological Malignancies in Kashmiri population. Asian Pac J Cancer Prev. 2019;20(12):3611-3615.
doi pubmed - Koczkodaj D, Muzyka-Kasietczuk J, Chocholska S, Podhorecka M. Prognostic significance of isochromosome 17q in hematologic malignancies. Oncotarget. 2021;12(7):708-718.
doi pubmed - Visconte V, Tabarroki A, Zhang L, Hasrouni E, Gerace C, Frum R, Advani AS, et al. Molecular characterization of myeloid neoplasms harboring isochromosome 17Q abnormality. Blood. 2013;122(21):2596.
doi - Kanagal-Shamanna R, Luthra R, Yin CC, Patel KP, Takahashi K, Lu X, Lee J, et al. Myeloid neoplasms with isolated isochromosome 17q demonstrate a high frequency of mutations in SETBP1, SRSF2, ASXL1 and NRAS. Oncotarget. 2016;7(12):14251-14258.
doi pubmed - Shin DY. TP53 mutation in acute myeloid leukemia: an old foe revisited. Cancers (Basel). 2023;15(19):4816.
doi pubmed - Welch JS. Patterns of mutations in TP53 mutated AML. Best Pract Res Clin Haematol. 2018;31(4):379-383.
doi pubmed - Zahid MF, Malik UA, Sohail M, Hassan IN, Ali S, Shaukat MHS. Cytogenetic abnormalities in myelodysplastic syndromes: an overview. Int J Hematol Oncol Stem Cell Res. 2017;11(3):231-239.
pubmed - Gurvich N, Voza F, Menendez S, Nimer S. Loss of L3MBTL1, a candidate 20Q12 tumor suppressor gene, leads to DNA damage. Blood. 2009;114(22):1974.
doi - Badar T, Nanaa A, Atallah E, Shallis RM, Guilherme SCC, Goldberg AD, Saliba AN, et al. Comparing venetoclax in combination with hypomethylating agents to hypomethylating agent-based therapies for treatment naive TP53-mutated acute myeloid leukemia: results from the Consortium on Myeloid Malignancies and Neoplastic Diseases (COMMAND). Blood Cancer J. 2024;14(1):32.
doi pubmed - Aldoss I, Zhang J, Pillai R, Shouse G, Sanchez JF, Mei M, Nakamura R, et al. Venetoclax and hypomethylating agents in TP53-mutated acute myeloid leukaemia. Br J Haematol. 2019;187(2):e45-e48.
doi pubmed - DiNardo CD, Jonas BA, Pullarkat V, Thirman MJ, Garcia JS, Wei AH, Konopleva M, et al. Azacitidine and venetoclax in previously untreated acute myeloid leukemia. N Engl J Med. 2020;383(7):617-629.
doi pubmed - Osman A. When are idiopathic and clonal cytopenias of unknown significance (ICUS or CCUS)? Hematology Am Soc Hematol Educ Program. 2021;2021(1):399-404.
doi pubmed - Xie Z, Nanaa A, Saliba AN, He R, Viswanatha D, Nguyen P, Jevremovic D, et al. Treatment outcome of clonal cytopenias of undetermined significance: a single-institution retrospective study. Blood Cancer J. 2021;11(3):43.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (CC BY 4.0), which permits unrestricted use, distribution, and reproduction in any medium, including commercial use, provided the original work is properly cited.
Journal of Medical Cases is published by Elmer Press Inc.