| Journal of Medical Cases, ISSN 1923-4155 print, 1923-4163 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Med Cases and Elmer Press Inc |
| Journal website https://jmc.elmerpub.com |
Case Report
Volume 17, Number 5, May 2026, pages 221-225

Successful Treatment of Overlapping Myelin Oligodendrocyte Glycoprotein Antibody-Associated Disease and Glial Fibrillary Acidic Protein Astrocytopathy With Plasma Exchange
Chisa Ashidaa, c, Tomohiro Suzukia, Jun Sawadaa, Ryo Yuzawab, Syuichi Tetsukaa, Koji Hosoyab, Tomoko Ogawaa, Ritsuo Hashimotoa
aDepartment of Neurology, International University of Health and Welfare Hospital, 537-3, Iguchi, Nasushiobara, Tochigi 329-2763, Japan
bDepartment of Nephrology, International University of Health and Welfare Hospital, 537-3, Iguchi, Nasushiobara, Tochigi 329-2763, Japan
cCorresponding Author: Chisa Ashida, Department of Neurology, International University of Health and Welfare Hospital, 537-3, Iguchi, Nasushiobara, Tochigi 329-2763, Japan
Manuscript submitted January 20, 2026, accepted February 27, 2026, published online March 27, 2026
Short title: MOGAD and GFAP Astrocytopathy Overlap Treated With PE
doi: https://doi.org/10.14740/jmc5290
| Abstract | ▴Top |
Myelin oligodendrocyte glycoprotein (MOG) antibody-associated disease (MOGAD) and glial fibrillary acidic protein (GFAP) astrocytopathy are distinct autoimmune inflammatory disorders of the central nervous system (CNS) that occasionally overlap. A 69-year-old man presented with acute gait disturbance, followed by progressive impairment of consciousness, myoclonus, cerebellar ataxia, and hyponatremia. Initial brain magnetic resonance imaging (MRI) revealed a high-intensity lesion in the left thalamus on diffusion-weighted imaging (DWI) and multiple white matter lesions on fluid-attenuated inversion recovery (FLAIR) imaging. While serum anti-MOG antibodies were positive, the clinical features, including progressive impairment of consciousness and hyponatremia, were atypical for MOGAD alone, leading to the identification of anti-GFAPα antibodies in the cerebrospinal fluid (CSF). High-dose corticosteroid therapy (three courses of intravenous methylprednisolone pulse) resulted in only partial clinical improvement. However, subsequent simple plasma exchange (PE) with fresh frozen plasma (FFP) led to marked neurological recovery. The mechanism of recovery is thought to involve the removal of both pathogenic antibodies and pro-inflammatory mediators. By day 82 of hospitalization, the patient was able to ambulate independently. This case suggests that in overlapping autoimmune syndromes, early escalation to PE should be considered when conventional steroid therapy is insufficient.
Keywords: MOGAD; GFAP astrocytopathy; Overlapping syndrome; Plasma exchange
| Introduction | ▴Top |
Myelin oligodendrocyte glycoprotein antibody–associated disease (MOGAD) and glial fibrillary acidic protein (GFAP) astrocytopathy are inflammatory central nervous system (CNS) disorders with overlapping clinical presentations, including encephalopathy, ataxia, and myelitis [1–3]. Although both conditions typically respond to corticosteroids [1–3], the coexistence of multiple autoimmune antibodies may contribute to more complex clinical presentations and heterogeneous treatment responses [3–5]. Distinguishing the predominant pathogenic antibody and identifying overlapping syndromes are essential for appropriate management [6].
Differential diagnosis of these conditions includes infectious meningoencephalitis, paraneoplastic neurological syndromes, and other autoimmune encephalitis such as anti-N-methyl-D-aspartate (NMDA) receptor encephalitis, as these conditions require fundamentally different diagnostic approaches and therapeutic considerations.
We report a case of overlapping MOGAD and GFAP astrocytopathy that was refractory to steroids but was successfully treated with simple plasma exchange (PE) with fresh frozen plasma (FFP).
| Case Report | ▴Top |
A 69-year-old man with no remarkable past medical history presented with acute gait disturbance. He had no headache or meningeal signs. Six days prior to onset, he had experienced symptoms of an upper respiratory tract infection. Upon admission, laboratory tests revealed hyponatremia (Na 124 mmol/L). Neurological examination showed impaired consciousness (Glasgow Coma Scale: E3V4M4; Japan Coma Scale: I-3∼II-10), hyperreflexia in the lower extremities, and gait instability primarily attributable to truncal ataxia and involuntary movements. No apparent muscle weakness or sensory deficits were observed, indicating that the gait disturbance at onset was predominantly cerebellar in origin. He subsequently developed postural tremor, myoclonus in the upper limbs, progressive cerebellar ataxia, and urinary retention. On day 11, the patient developed respiratory failure and required endotracheal intubation and mechanical ventilation.
Brain magnetic resonance imaging (MRI) on admission showed a high-intensity lesion in the left thalamus on diffusion-weighted imaging (DWI) with a corresponding low-intensity lesion on the apparent diffusion coefficient (ADC) map, along with multiple spotty white matter lesions on fluid-attenuated inversion recovery (FLAIR) (Fig. 1). Magnetic resonance angiography (MRA) showed no significant stenosis. Cerebrospinal fluid (CSF) analysis revealed pleocytosis (97/mm3), protein elevation (153 mg/dL), and positive oligoclonal bands (six bands). The immunoglobulin G (IgG) index was 0.959. Serum anti-MOG antibodies were positive (titer 1:16, Cell-based assay), whereas anti-aquaporin-4 (AQP4), anti-NMDA, and various paraneoplastic antibodies were negative (Table 1).
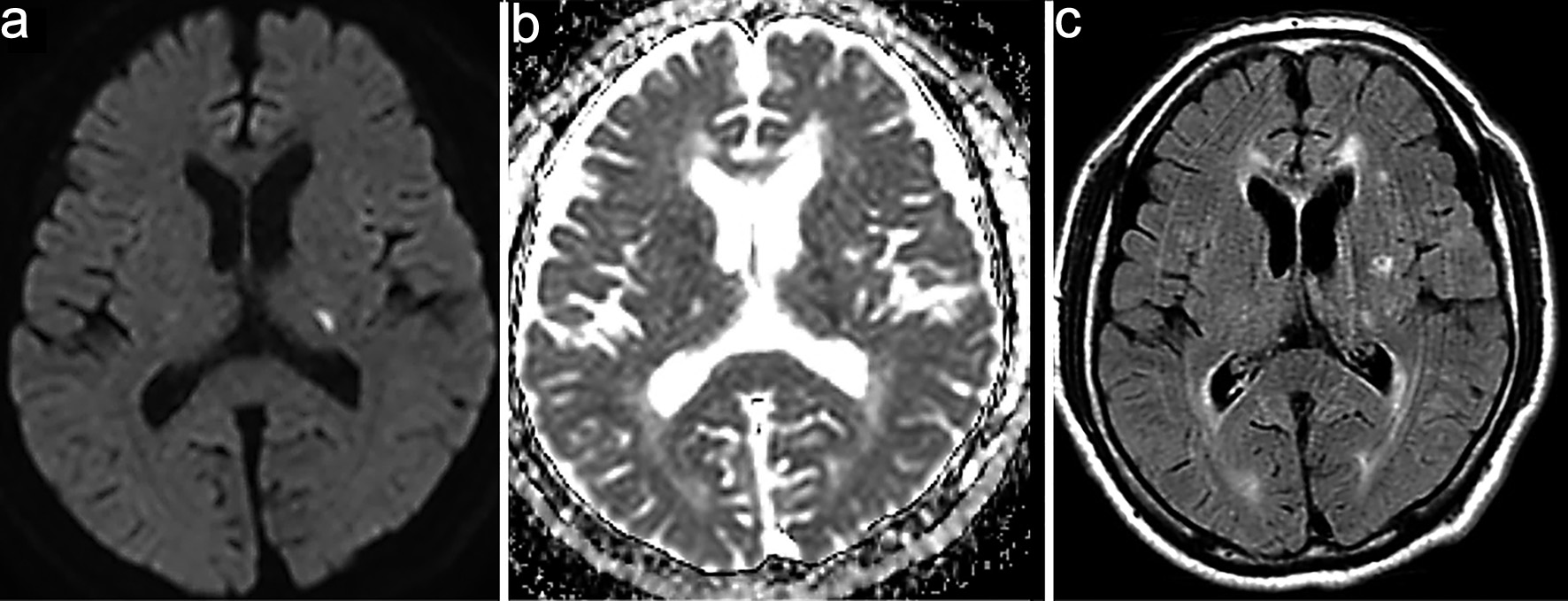 Click for large image | Figure 1. Magnetic resonance imaging (MRI) on admission. (a) High-intensity lesion in the left thalamus on diffusion-weighted imaging (DWI). (b) Corresponding low-intensity lesion on the apparent diffusion coefficient (ADC) map. (c) Fluid-attenuated inversion recovery (FLAIR) images demonstrating multiple spotty hyperintense white matter lesions. |
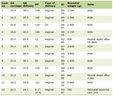 Click to view | Table 1. Laboratory Findings |
Although the positive results for anti-MOG antibodies led us to consider MOGAD as the initial diagnosis, the patient’s clinical course, including progressive impairment of consciousness and hyponatremia, was atypical. Serial follow-up MRI (DWI) revealed new small hyperintense lesions appearing sequentially in the left corona radiata and near the occipital horn of the left lateral ventricle (Fig. 2). On day 11, contrast-enhanced MRI showed serpiginous perivascular enhancement in the basal ganglia along with leptomeningeal enhancement (Fig. 3); however, these findings were not clearly consistent with the typical radial periventricular enhancement pattern reported in GFAP astrocytopathy. Given these atypical clinical and radiological findings, additional autoimmune antibody testing was performed, which confirmed the presence of anti-GFAPα antibodies in the CSF (Cell-based assay). Spinal cord imaging was performed and revealed no abnormalities. The patient had no visual symptoms, including visual disturbance, ocular pain, or visual field impairment. Dedicated optic nerve MRI was not obtained; however, the patient was evaluated by ophthalmology, which identified perivascular inflammation around the right optic nerve. Whole-body computed tomography (CT) screening and tumor markers revealed no malignancy.
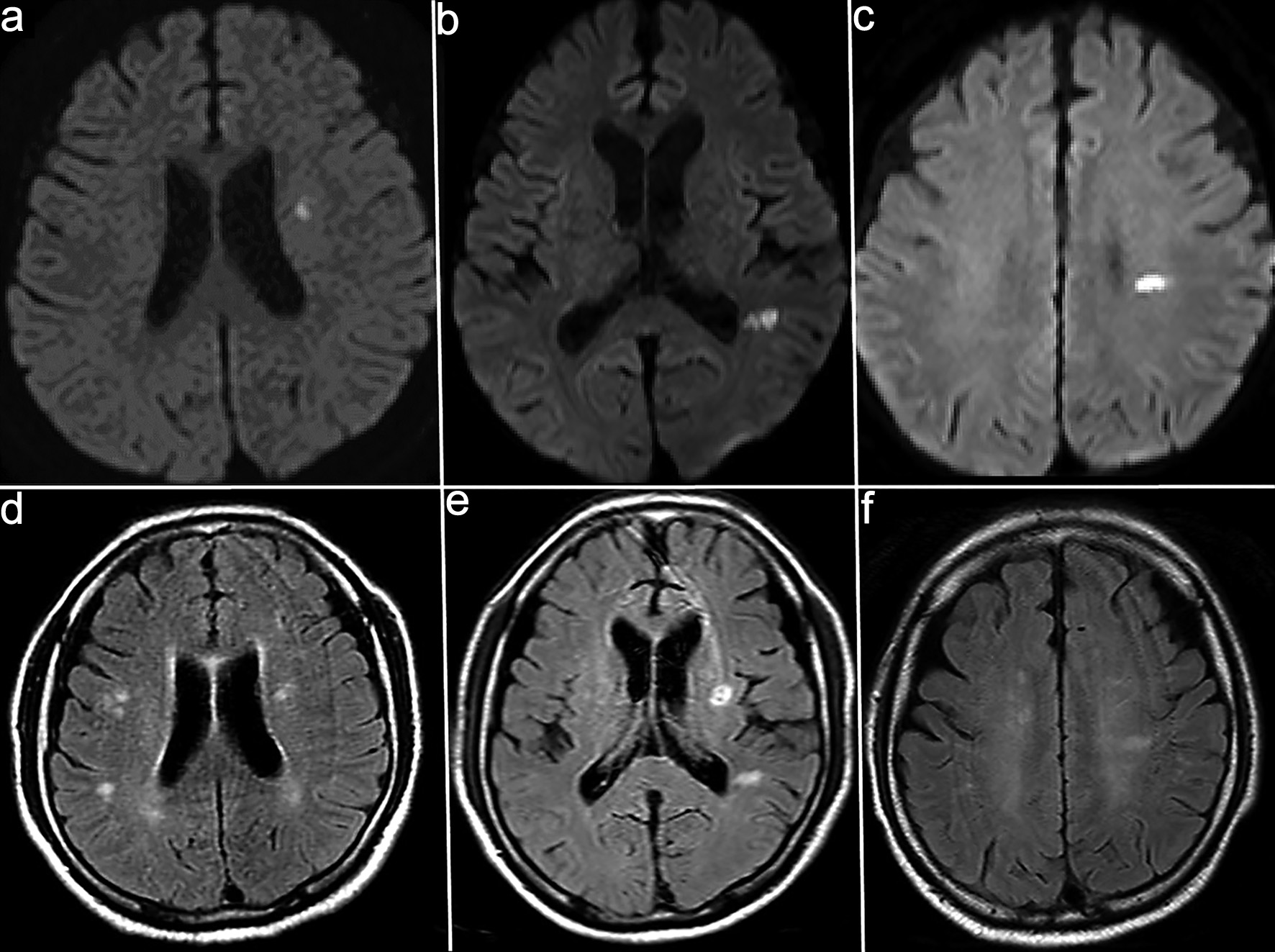 Click for large image | Figure 2. Serial diffusion-weighted imaging (DWI) showing the appearance of new lesions. (a–c) Follow-up magnetic resonance imaging (MRI) on day 7 (a), day 20 (b), and day 35 (c) shows small hyperintense lesions appearing sequentially in the left corona radiata and near the occipital left horn of the lateral ventricle. (d–f) Corresponding fluid-attenuated inversion recovery (FLAIR) images acquired at the same time points (a–d, b–e, and c–f pairs) demonstrate hyperintense white matter lesions. |
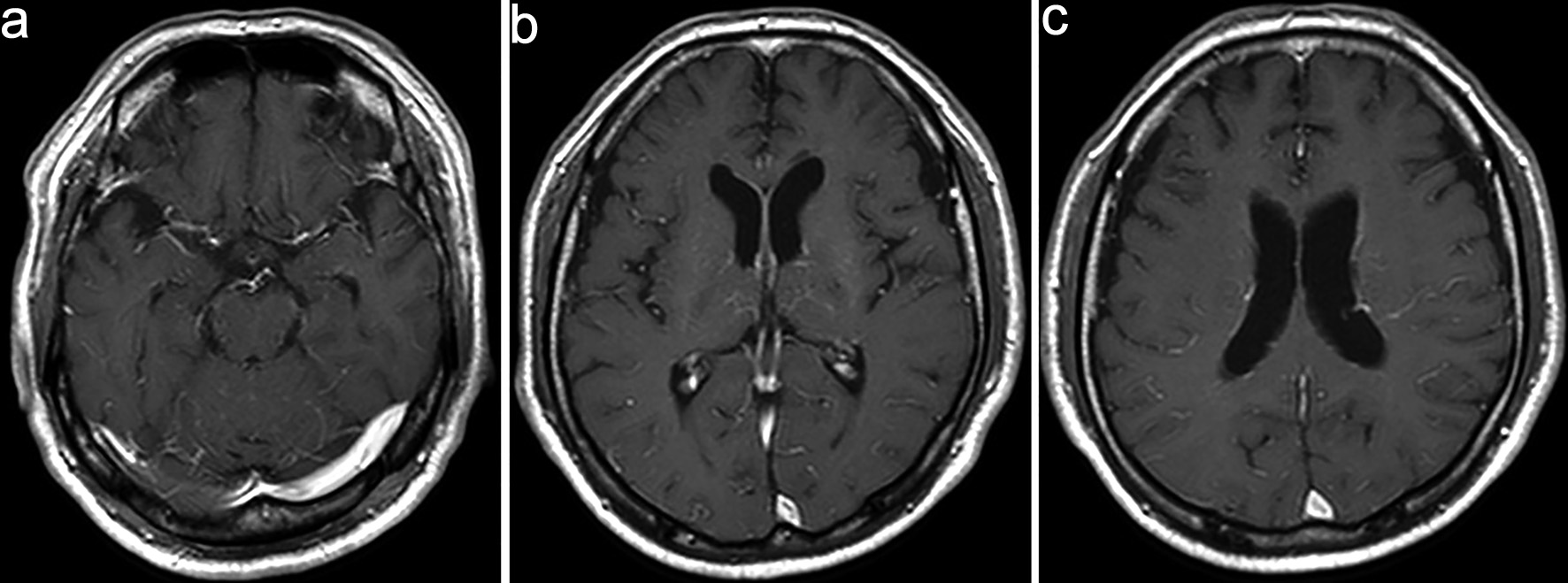 Click for large image | Figure 3. Contrast-enhanced magnetic resonance imaging (MRI) on day 11. (a–c) Contrast-enhanced MRI shows serpiginous perivascular enhancement in the basal ganglia along with leptomeningeal enhancement. |
The patient was treated with three courses of intravenous methylprednisolone pulse (IVMP) therapy (1,000 mg/day for 3 days per course). Following IVMP, serum sodium levels normalized and respiratory status improved, allowing successful weaning from mechanical ventilation. However, the response was partial, and impaired consciousness (Glasgow Coma Scale: E3V4M6; Japan Coma Scale: II-10), myoclonus, tremor, and severe cerebellar ataxia persisted. Simple PE with FFP (three sessions) was then initiated on days 40, 45, and 47, which resulted in significant neurological improvement, including restoration of clear consciousness (Glasgow Coma Scale: E4V5M6; Japan Coma Scale: 0). Myoclonus and postural tremor resolved, and cerebellar ataxia markedly improved, enabling the patient to sit and stand independently and eventually ambulate without assistance. However, urinary retention persisted and continued to require management. Following PE, the CSF cell count significantly improved from 235 to 10/mm3, and no new lesions were detected on brain MRI, and previously observed enhancement resolved; therefore, PE was discontinued after three sessions. He was discharged on day 82 with the ability to ambulate independently while continuing oral prednisolone (20 mg/day).
| Discussion | ▴Top |
The coexistence of multiple autoantibodies, termed “overlapping syndrome,” has been increasingly reported [7]. Among these, the overlap of anti-MOG antibodies and anti-GFAP antibodies represents a unique clinical entity where two distinct pathogenic processes, demyelination and astrocytopathy, occur simultaneously.
In our patient, several clinical clues suggested that GFAP astrocytopathy was a major contributor despite the initial detection of MOG antibodies. First, the progressive encephalopathy, myoclonus, cerebellar ataxia, and urinary retention are highly characteristic of GFAP astrocytopathy [8]. While MOGAD can present with acute disseminated encephalomyelitis (ADEM)-like features [2], the severity of the consciousness impairment was more suggestive of an astrocytopathic process [9]. Second, the marked hyponatremia (Na 124 mmol/L) is a well-documented feature of GFAP astrocytopathy [8], likely due to the involvement of the hypothalamus, whereas it is uncommon in isolated MOGAD. Although the patient fulfilled the 2023 international diagnostic criteria for MOGAD [2], several clinical red flags including poor responsiveness to corticosteroid therapy and oligoclonal band positivity, indicated an atypical presentation for MOGAD, supporting GFAP astrocytopathy as the predominant pathogenic process.
Although both MOGAD and GFAP astrocytopathy are generally steroid-responsive and previously reported cases of overlapping MOGAD and GFAP astrocytopathy have shown favorable outcomes [3, 4], our patient exhibited a severe clinical course requiring multimodal immunotherapy. In the present case, high-dose corticosteroid therapy resulted in partial clinical improvement, including normalization of hyponatremia and recovery of respiratory function, allowing discontinuation of mechanical ventilation. However, impaired consciousness, myoclonus, tremor, and cerebellar ataxia persisted despite steroid therapy, indicating incomplete treatment response. Notably, most reported overlap cases recovered well after immunotherapy, whereas the present case required mechanical ventilation and escalation to PE. One possible explanation is advanced age, as older patients may be more susceptible to severe disease progression and poorer treatment responsiveness. Therefore, age and rapid clinical deterioration may represent important factors prompting early therapeutic escalation in overlapping autoimmune CNS syndromes.
The pathophysiology of this overlapping syndrome remains to be fully elucidated. However, it may involve “epitope spreading,” a phenomenon where tissue damage caused by the primary autoimmune attack releases secondary antigens, triggering a secondary immune response [6]. This mechanism could explain the simultaneous presence of anti-GFAP and anti-MOG antibodies.
Subsequent PE resulted in marked improvement of these residual neurological symptoms, ultimately enabling independent ambulation. This stepwise therapeutic response suggests that corticosteroids were effective in controlling systemic inflammatory activity, whereas PE played a critical role in removing circulating pathogenic factors contributing to persistent neurological dysfunction. PE is thought to exert therapeutic effects not only through the removal of pathogenic autoantibodies but also by eliminating circulating pro-inflammatory cytokines and immune complexes that sustain CNS inflammation [10]. Therefore, in overlapping autoimmune CNS syndromes, escalation to PE should be considered when neurological recovery remains incomplete after corticosteroid therapy.
Furthermore, long-term management requires careful monitoring. While GFAP astrocytopathy may follow a monophasic course, the presence of MOG antibodies increases the likelihood of a relapsing-remitting course [4, 5]. Therefore, maintaining moderate-dose oral prednisolone after discharge was deemed necessary to prevent recurrence [4].
Conclusion
This case illustrates that overlapping autoimmune CNS disorders should be suspected when clinical manifestations, such as severe encephalopathy or hyponatremia, cannot be fully explained by a single antibody. Early identification of anti-MOG antibodies and anti-GFAP antibodies and prompt initiation of PE may be considered in achieving favorable outcomes in steroid-refractory cases.
Acknowledgments
We thank the patient for his cooperation in the preparation of this case report.
Financial Disclosure
None to declare.
Conflict of Interest
The authors declare that there is no conflict of interest relevant to this article.
Informed Consent
Written informed consent was obtained from the patient’s family for publication of this case report.
Author Contributions
Study design was conducted by CA and TS. PE was performed by RY and KH. Patient management was conducted by CA, JS, TS, ST, RH, and TO. CA wrote the manuscript which was revised and approved by all authors.
Data Availability
The data supporting the findings of this study are available from the corresponding author upon reasonable request.
Abbreviations
ADC: apparent diffusion coefficient; ADEM: acute disseminated encephalomyelitis; AMPH: amphiphysin; ANA: antinuclear antibody; AQP4: aquaporin-4; APS: antiphospholipid syndrome; CASPR2: contactin-associated protein-like 2; CL-β2GP1: cardiolipin–β2 glycoprotein I; CNS: central nervous system; CSF: cerebrospinal fluid; CV2 (CRMP5): collapsin response mediator protein 5; dsDNA: double-stranded DNA; DWI: diffusion-weighted imaging; FLAIR: fluid-attenuated inversion recovery; GAD65: glutamic acid decarboxylase 65; GFAP: glial fibrillary acidic protein; HIV: human immunodeficiency virus; IgG: immunoglobulin G; IgLON5: immunoglobulin-like cell adhesion molecule LON5; IVMP: intravenous methylprednisolone pulse; LGI1: leucine-rich glioma-inactivated 1; MOG: myelin oligodendrocyte glycoprotein; MOGAD: myelin oligodendrocyte glycoprotein antibody–associated disease; MRI: magnetic resonance imaging; MRA: magnetic resonance angiography; NMDA: N-methyl-D-aspartate; PE: plasma exchange; PNMA2 (Ma2): paraneoplastic Ma antigen 2; Recoverin: recoverin protein; Ri (ANNA-2): Ri antigen; SOX1: SRY-box transcription factor 1; SSA: Sjogren’s syndrome–related antigen A; SSB: Sjogren’s syndrome–related antigen B; Titin: titin protein; TPO: thyroid peroxidase; Tr (DNER): delta/notch-like epidermal growth factor–related receptor; Yo (PCA-1): Yo antigen; Zic4: zinc finger protein of the cerebellum 4
| References | ▴Top |
- Fang B, McKeon A, Hinson SR, Kryzer TJ, Pittock SJ, Aksamit AJ, Lennon VA. Autoimmune glial fibrillary acidic protein astrocytopathy: a novel meningoencephalomyelitis. JAMA Neurol. 2016;73(11):1297-1307.
doi pubmed - Japanese Society of Neurology. Guidelines for multiple sclerosis and neuromyelitis optica spectrum disorders 2023. Tokyo: Igaku-Shoin; 2023. https://www.neurology-jp.org/guidelinem/pdf/koukasyo_nmosd_2023.pdf.
- Shan F, Long Y, Qiu W. Autoimmune glial fibrillary acidic protein astrocytopathy: a review of the literature. Front Immunol. 2018;9:2802.
doi pubmed - Li Q, Chen X. Clinical characteristics of patients with GFAP-IgG coexisting with AQP4-IgG or MOG-IgG. Front Immunol. 2025;16:1610486.
doi pubmed - Dubey D, Hinson SR, Jolliffe EA, Zekeridou A, Flanagan EP, Pittock SJ, Basal E, et al. Autoimmune GFAP astrocytopathy: prospective evaluation of 90 patients in 1 year. J Neuroimmunol. 2018;321:157-163.
doi pubmed - Zhao C, Liu P, Zhao D, Ding J, Zhang G, Li H, Guo J. Coexistence of myelin oligodendrocyte glycoprotein immunoglobulin g and neuronal or glial antibodies in the central nervous system: a systematic review. Brain Sci. 2022;12(8):850651.
doi pubmed - Yang X, Xu H, Ding M, Huang Q, Chen B, Yang H, Liu T, et al. Overlapping autoimmune syndromes in patients with glial fibrillary acidic protein antibodies. Front Neurol. 2018;9:251.
doi pubmed - Kimura A, Takekoshi A, Yoshikura N, Hayashi Y, Shimohata T. Clinical characteristics of autoimmune GFAP astrocytopathy. J Neuroimmunol. 2019;332:91-98.
doi pubmed - Gklinos P, Athanasopoulos F, Giatrakou V, Arkoudis NA, Pournara D, Giagkou E, Tountopoulou A, et al. Unveiling GFAP astrocytopathy: insights from case studies and a comprehensive review of the literature. Antibodies (Basel). 2024;13(4):79.
doi pubmed - Tonev DG, Momchilova AB. Therapeutic plasma exchange in certain immune-mediated neurological disorders: focus on a novel nanomembrane-based technology. Biomedicines. 2023;11(2):328.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (CC BY 4.0), which permits unrestricted use, distribution, and reproduction in any medium, including commercial use, provided the original work is properly cited.
Journal of Medical Cases is published by Elmer Press Inc.