| Journal of Medical Cases, ISSN 1923-4155 print, 1923-4163 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Med Cases and Elmer Press Inc |
| Journal website https://jmc.elmerpub.com |
Case Report
Volume 000, Number 000, October 2025, pages 000-000

Perioperative Care of a Child With Combined Oxidative Phosphorylation Deficiency 6: Total Intravenous Anesthesia With Remimazolam
Ifeoluwa C. Olakunlea, Joseph D. Tobiasa, b, c
aDepartment of Anesthesiology and Pain Medicine, Nationwide Children’s Hospital, Columbus, OH, USA
bDepartment of Anesthesiology and Pain Medicine, The Ohio State University College of Medicine, Columbus, OH, USA
cCorresponding Author: Joseph D. Tobias, Department of Anesthesiology and Pain Medicine, Nationwide Children’s Hospital, Columbus, OH 43205, USA
Manuscript submitted July 31, 2025, accepted September 17, 2025, published online October 10, 2025
Short title: Anesthesia and COXPD6
doi: https://doi.org/10.14740/jmc5179
| Abstract | ▴Top |
Combined oxidative phosphorylation deficiency 6 (COXPD6) is a severe mitochondrial encephalomyopathy resulting from a mutation in the X-linked apoptosis-inducing factor mitochondrion-associated 1 (AIFM1) gene. The AIFM1 gene located on chromosome Xq26.1, encodes apoptosis inducing factor (AIF), a flavin adenine dinucleotide (FAD)-dependent nicotinamide adenine dinucleotide (NADH) oxidoreductase, which is involved in the process of oxidative phosphorylation and mitochondrial-derived programmed cell death in the nucleus. COXPD6 patients have significant end-organ involvement of the central nervous, peripheral nervous, respiratory, and gastrointestinal systems, manifested by seizures, hypotonia, psychomotor delay, muscle weakness, and wasting. We present an 11-year-old child with AIFM1-related COXPD6 who underwent posterior spinal fusion for progressive neuromuscular kyphoscoliosis. We explore the genetic history of this mitochondrial disorder, review a detailed anesthetic approach to perioperative management including use of the novel benzodiazepine, remimazolam, and discuss anesthetic considerations in these patients.
Keywords: AIFM1 gene; Mitochondrial disease; AIFM1-related combined oxidative phosphorylation deficiency 6; Pediatric anesthesiology
| Introduction | ▴Top |
There is a dual genetic control of mitochondria, with maternally inherited mitochondrial DNA (mtDNA), which encodes 37 genes, and the nuclear genome (nDNA) of the cell, which encodes additional genetic material encoding proteins that contribute to its structure and function. Consequently, mitochondrial diseases can occur from varying genetic mutations involving the mitochondrial or nuclear genome leading to disruptions in mitochondrial function and processes [1-3]. Given their primary role in the production of adenosine triphosphate (ATP) and generation of the energy required for cellular homeostasis, defects in mitochondrial function can have varied end-organ manifestations with predominant involvement of high energy tissues, including the central nervous, cardiac, and skeletal muscles systems. Clinical signs and symptoms range from easily identifiable mitochondrial syndromes such as mitochondrial myopathies, neuropathies, and encephalomyopathies to complex multi-system diseases [4]. Advances in the understanding of the cellular process of mitochondria and laboratory assays have led to the identification of several enzymatic deficiencies resulting in mitochondrial disorders.
Combined oxidative phosphorylation deficiency 6 (COXPD6) (Online Mendelian Inheritance in Man (OMIM) #300816) is a severe mitochondrial encephalomyopathy caused by a hemizygous mutation in the X-linked apoptosis-inducing factor mitochondrion-associated 1 (AIFM1) gene. The AIFM1 gene located on chromosome Xq26.1, encodes apoptosis inducing factor (AIF), a flavin adenine dinucleotide (FAD)-dependent nicotinamide adenine dinucleotide (NADH) oxidoreductase that plays a role in oxidative phosphorylation (OXPHOS) and mitochondrial-derived programmed cell death in the nucleus [5, 6]. The AIFM1 gene mutation has several diseases variants including Charcot-Marie-Tooth disease type 4 (CMTX4, Cowchock syndrome), X-linked deafness-5, and X-linked spondyloepimetaphyseal dysplasia with hypomyelinating leukodystrophy. Among the AIFM1-related diseases, COXPD6 (the specific disorder noted in our patient) is a more severe disorder, involving both the central and peripheral nervous systems, with severe phenotypic manifestation in each. Respiratory and gastrointestinal system involvement can also occur. The implicated AIFM1 gene mutation results in a defect in mitochondrial-driven OXPHOS, resulting in seizures, hypotonia, psychomotor delay, muscle weakness, and wasting [5-7].
We present an 11-year-old child with AIFM1-related COXPD6 (CMTX4), who underwent posterior spinal fusion for progressive neuromuscular kyphoscoliosis. We explored the genetic history of this mitochondrial disorder and outlined a detailed anesthetic approach to perioperative management including the use of the novel benzodiazepine, remimazolam, in these patients.
| Case Report | ▴Top |
Preoperative history and clinical course
The patient was an 11-year-old boy, weighing 41 kg, born at term via vaginal delivery. The antenatal course, neonatal history, and early infancy were uneventful with normal psychomotor development. At 12 - 15 months of age, his mother reported the onset of an abnormal gait which progressed to involve tremors, extremity stiffness, frequent falls, and behavioral disturbances. Continued worsening symptoms prompted referral to a neurologist by his primary care physician at 2 years of age. At the time of his first visit, developmental history was significant for language and social delays; however, cognitive and motor development appeared normal. The initial diagnostic workup included brain magnetic resonance imaging (MRI), electroencephalography (EEG), and neuronal ceroid lipofuscinosis enzymatic testing, all of which were all normal. A working diagnosis of myoclonic disorder and attention deficit hyperactivity disorder (ADHD) was made. Chromosomal microarrays with reflex karyotype, and myotonic dystonia epsilon sarcoglycan (SGCE) gene mutation testing were negative. He continued to have severe symptoms with frequent falls, necessitating further referral to establish a definitive diagnosis. At 3 years old, he was referred to the genetics clinic for consultation, and an expanded metabolic and genetic workup was done. Testing for fragile X syndrome, spinocerebellar ataxia, metabolic screening, and comprehensive ataxia genetic panel were all normal and non-diagnostic. Whole exome sequencing genetic testing revealed a hemizygous AIFM1 gene mutation associated with X-linked recessive CMTX4. His clinical presentation evolved with recurrent generalized seizures and myoclonic movements indicative of encephalopathy. This led to a definitive diagnosis of AIFM1-related combined OXPHOS deficiency. Since diagnosis, his clinical course has been marked by multiple hospitalizations, hearing impairment, speech and language disorders, behavioral disorders, feeding disorders, obstructive sleep apnea (OSA), restrictive lung disease, spinal instability with need for assisted mobility devices, and multi-system involvement requiring multi-disciplinary care. His past surgical history was significant for surgical correction of an incomplete circumcision with penile adhesions at 19 months of age, as well as bilateral cochlear implant and placement of a gastrostomy (G)-tube for feeding at 5 years of age. Due to his worsening neuromuscular kyphoscoliosis, he was scheduled for a posterior spinal instrumentation of the T3-L3 vertebrae segments.
Preoperative evaluation
At the time of procedure, the patient had a feeding G-tube in place, and his medication regimen included clonidine, clonazepam, trihexyphenidyl, gabapentin, chlorpromazine, melatonin, propranolol, vitamins supplementation, inhaled bronchodilators, nocturnal continuous airway positive pressure (6 cm H2O) for OSA and home oxygen therapy as needed. Preoperative coagulation function, complete blood count, serum electrolytes, and renal function test were normal. His weight was 41 kg. Preoperative vital signs were unremarkable. On physical examination, the Mallampati class was unable to be assessed while the thyromental distance was more than three finger-breadths. Cardiac, craniofacial, and respiratory examinations were otherwise unremarkable. His American Society of Anesthesiologist (ASA) physical status classification was 4.
Intraoperative course
The patient was kept NPO for 6 h and, following premedication with oral midazolam and aprepitant, and was transported to the operating room. Intraoperative medications and their indication are listed in Table 1. Routine ASA monitors were applied, and anesthesia was induced. The trachea was intubated with a 6.0-mm cuffed endotracheal tube, and a bispectral index (BIS) monitor was placed to monitor the depth of anesthesia. Two additional peripheral intravenous cannulas and a left radial arterial cannula were inserted. The patient was positioned prone on the Jackson table with padding of pressure points. The room was warmed, and a forced hair warming device was placed to maintain normothermia. Maintenance anesthesia and other intraoperative medications are outlined in Table 1. Medications were adjusted to maintain the BIS at 50 - 60 and mean arterial pressure (MAP) at 50 - 65 mm Hg. There were no significant intraoperative concerns. The total anesthesia time was 5 h. Intraoperative fluids included Normosol-R and buffered saline 2% for a total volume of 1,300 mL. Estimated blood loss was 100 mL with urine output of 670 mL. At the completion of the procedure, the anesthetic agents were discontinued, sugammadex (2 mg/kg) was administered to reverse residual neuromuscular blockade, and the patient was turned supine. His trachea was successfully extubated once awake. Additional bolus doses of fentanyl (1.2 µg/kg) for pain control and dexmedetomidine (1 µg/kg) to control agitation on emergence were administered.
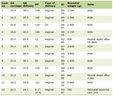 Click to view | Table 1. Intraoperative Medications and Indications |
Postoperative course
The patient was transported to the post-anesthesia care unit (PACU) and thereafter admitted to the pediatric intensive care unit (PICU) for immediate postoperative monitoring. He had a multimodal pain regimen with IV hydromorphone administered via patient-controlled analgesia, scheduled IV acetaminophen, scheduled IV ketorolac, lidocaine infusion, and diazepam as needed. Routine chest physiotherapies were initiated, and he was placed on his usual home continuous positive airway pressure (6 cm H2O) at night. Routine home medications including clonidine, gabapentin, clonazepam, trihexyphenidyl and chlorpromazine were resumed on postoperative day (POD) 1. On POD 1, he was transferred to the inpatient ward. There was some leakage noted around the G-tube. The postoperative course was complicated by an ileus requiring that he be placed NPO, respiratory insufficiency requiring supplemental oxygen, and concerns for benzodiazepine withdrawal from his usual home medication regimen. Ongoing oxygen desaturation resulted in the need to call the rapid response team, and he was transferred back to the PICU for hemodynamic and respiratory monitoring. A chest radiograph showed increased left lower lobe atelectasis and pneumonia. A repeat abdominal radiograph revealed mild improvement of ileus. A nasal swab for respiratory viruses (polymerase chain reaction testing) was positive for non-coronavirus disease 2019 (COVID-19) coronavirus. Laboratory evaluation revealed that creatinine phosphokinase was elevated at 10,109 U/L (reference less than 430 U/L). The latter was concerning for rhabdomyolysis secondary to dystonia precipitated by benzodiazepine withdrawal. Ongoing PICU care with a focus on his respiratory status and control of postoperative agitation with diazepam and haloperidol resulted in an overall improvement in his clinical status. On POD 4, he was stable on room air, feeds were slowly restarted, and routine oral medications were resumed via the G-tube. Laboratory values also showed a down-trending of serum creatinine phosphokinase levels. He was transferred back to the inpatient ward. Ongoing ileus required placement of a jejunostomy (J)-tube through the gastrostomy site for enteral feeding. The postoperative course was further complicated by deep and superficial venous thrombosis involving the subclavian, cephalic, and basilic veins on the right with superimposed cellulitis, tenosynovitis, and osteomyelitis. Blood culture yielded no growth. Due to the venous thrombosis, enoxaparin was started. By POD 42, his clinical condition had significantly improved, and his care was transferred to physical medicine for inpatient rehabilitation. He was successfully discharged home on POD 82 with follow-up at the orthopedic surgery, behavioral health, pulmonary, nephrology, and complex care clinics.
| Discussion | ▴Top |
The current case report and following discussion outline pertinent concerns when providing perioperative management of a patient with COXPD6, a severe mitochondrial encephalomyopathy. As with other disorders involving mitochondrial function, the primary end-organ involvement includes the central nervous system (CNS, seizures, cognitive delay), the skeletal musculature (hypotonia), and myocardial function. Of note, theoretical concerns have been expressed regarding the potential adverse effects of both volatile anesthetic agents and propofol on mitochondrial function.
AIF is a 67-kDa, AIFM1 gene-encoded flavoprotein that plays a crucial role in mitochondrial metabolism, participating in the electron transport chain (ETC) assembly where it regulates mitochondrial OXPHOS and ATP production [7, 8]. Consequently, a decrease or absence in AIF activity can cause complex defects and a reduction in the nuclear-encoded protein subunits of complex I, and to a lesser extent complex III and IV, resulting in OXPHOS dysfunction. In addition, the mitochondrial respiratory chain protein complexes not only catalyze OXPHOS but also play a role in reactive oxygen species (ROS) generation. Therefore, the ETC dysfunction produces energy failure and additional biochemical effects, including increased production of ROS [7, 9-11].
Mutations in the AIFM1 gene may be responsible for a range of mitochondrial disorders. The growing number of novel AIFM1 mutations, their associated AIF variants and varying clinical phenotypes are widening the clinical spectrum of AIF-related disorders and making their categorization a challenge. COXPD6 is one of the disease categories associated with the AIFM1 mutation. It is caused by a hemizygous AIFM1 mutation that impairs OXPHOS and phenotypically presents with severe mitochondrial encephalomyopathy [12-15]. Reported cases of COXPD6 have similarities in organ system involvement, with variations noted in severity, age of onset, and outcomes.
The muscles, central and peripheral nervous system are involved early with symptoms of hypotonia, absent or reduced reflexes, involuntary movements, seizures, psychomotor regression, progressive muscle weakness, muscle atrophy and abnormal basal ganglia signals on MRI. Hearing loss and ocular involvements can also be present. Gastrointestinal and respiratory involvements include swallowing difficulties, cyanosis, respiratory muscle weakness, and failure requiring ventilator support [6, 10, 15-17]. Anecdotal experience from two case reports have noted metabolic acidosis and increased lactate in the serum and cerebrospinal fluid [10, 16]. Other diagnostic studies such as muscle and skin biopsies, and Western blot analysis show decreased respiratory chain complexes activities and decreased levels of the AIFM1 protein. Genomic analysis using whole exome sequencing and Sanger sequencing can be done to make a definitive diagnosis and identify the implicated mutation [16, 17].
Considering the organ system involvement in COXPD6, affected children often require diagnostic and therapeutic interventions, such as radiologic imaging, tracheostomy, feeding tube placement or orthopedic procedures, thereby necessitating anesthetic care. As with all anesthetic care, the approach to these patients begins with a thorough preoperative history, examination and identification of associated comorbid conditions. One of the primary challenges in these patients is choice of anesthetic agent, as there are theoretical concerns regarding the use of either the volatile anesthetic agents or propofol, given their impact on mitochondrial processes [1, 18-20, 21-23]. Both propofol and volatile anesthetic agents may depress OXPHOS. Volatile anesthetic agents suppress OXPHOS by impacting complex I, coenzyme Q, and complex V of the mitochondrial respiratory chain. Patients with mitochondrial myopathies may have a heightened sensitivity to the effect of the volatile anesthetic agents on myocardial and CNS function along with a potential association, dependent on the specific defect, with malignant hyperthermia [1, 24]. Propofol acts on complex I and IV inhibiting mitochondrial function and uncoupling OXPHOS of fatty acids. This inhibition of mitochondrial function may lead to the classic findings of propofol infusion syndrome including renal failure, rhabdomyolysis, myocardial dysfunction, and metabolic acidosis.
With these concerns in mind, we chose to use total intravenous anesthesia (TIVA) with remimazolam and remifentanil. Following Food and Drug Administration (FDA) approval for use in adults in 2020, initial clinical trials have demonstrated the efficacy of remimazolam for sedation of adults during invasive procedures such as gastrointestinal endoscopy and bronchoscopy [25]. These trials have demonstrated its efficacy for procedural sedation as well as an acceptable safety profile with limited effects on hemodynamic function, lack of pain with IV administration, reduction of post-procedure nausea and vomiting (PONV), and a rapid return to baseline neurologic function. Similar to other benzodiazepines, remimazolam provides sedation, amnesia, and anxiolysis through the gamma-aminobutyric acid (GABA) system. As an ester-based medication, it is hydrolyzed quickly with a more rapid offset than midazolam and a limited context-sensitive half-life. Although not FDA-approved for use in children, initial anecdotal experience and trials (prospective and retrospective) have demonstrated its efficacy in a wide range of clinical scenarios in pediatric patients [25]. Anecdotal experience has demonstrated the safe and effective use of remimazolam in patients with mitochondrial dysfunction [26-29]. Given these concerns, we chose to avoid the use of volatile anesthetic agents and propofol and proceed with TIVA bolus doses of remimazolam, supplemented by fentanyl for anesthetic induction. This was followed by maintenance anesthesia with remimazolam, remifentanil, and lidocaine infusions. The remimazolam infusion was titrated by depth of anesthesia monitoring with the BIS.
Additional clinical considerations regarding anesthetic care involve the specific end-organ involvement. Cardiac considerations in children with COXPD6 include cardiac conduction abnormalities and primary myocardial involvement such as hypertrophic cardiomyopathy [6]. Routine preoperative cardiac evaluation should include an electrocardiogram and echocardiogram. In addition to its other beneficial properties, preliminary clinical experience has demonstrated a limited impact of remimazolam on myocardial conduction and contractility. Given the planned surgical procedure, continuous monitoring of arterial blood pressure was instituted in addition to standard ASA monitoring in our patient. With the potential for impaired mitochondrial function and its impact on lactate metabolism, lactate-containing fluids were avoided. Intraoperative maintenance and resuscitation fluids included our standard mix of Normosol-R, which contains gluconate and acetate as buffers, and buffered 2% saline containing acetate, for a total volume of 1,300 mL. Depending on the specific enzymatic defect, altered glucose metabolism with diabetes mellitus and hyperglycemia or hypoglycemia may be seen. Hyperglycemia and diabetes mellitus are generally seen late in the course of the disease processes. Hypoglycemia is most commonly encountered during acute illnesses or prolonged fasting without the administration of exogenous glucose. Given the potential for hypoglycemia, NPO times should be limited and periodic monitoring of serum glucose should be monitored intraoperatively for prolonged procedures.
Additional significant comorbid involvement in these patients includes hypotonia which poses a risk for perioperative aspiration, upper airway obstruction, and hypoventilation leading to respiratory failure. These respiratory concerns may be compounded by poor cough effort, chronic aspiration or recurrent pneumonia. Given these concerns, whenever feasible, short-acting anesthetic agents are recommended to allow for rapid awakening and limited impact on postoperative upper airway and respiratory function. Per our usual practice, following tracheal extubation, our patient was placed on noninvasive ventilatory support with biphasic positive airway pressure (BiPAP) along with aggressive chest physiotherapy to facilitate secretion management. Although significant postoperative respiratory issues were noted which resulted in a prolonged recovery and extended during of hospital stay, this was precipitated by benzodiazepine withdrawal and a concomitant viral respiratory infection. His respiratory status returned to baseline once those underlying pathologies were addressed. Given the potential for postoperative respiratory issues, continuous postoperative monitoring of respiratory function may be indicated following prolonged surgical procedures.
Hypotonia may impact the choice of neuromuscular blocking agents (NMBAs). Patients with pre-existing hypotonia related to mitochondrial disorders may be sensitive to the effects of non-depolarizing NMBAs [30]. Although we chose to use rocuronium to facilitate endotracheal intubation, even in the setting of hypotonia, sugammadex offers the potential to effectively reverse significant residual neuromuscular blockade in patients with neuromyopathic conditions [31]. While the data are limited, the associated myopathic condition would be a contraindication to the use succinylcholine given the risks for hyperkalemia [32]. In our patient, we chose rocuronium for intraoperative neuromuscular blockade with sugammadex for reversal. Additional CNS concerns include the presence of seizures. Preoperative management to limit perioperative seizures includes optimizing anticonvulsant medications prior to the surgical procedure and continuation of routine anticonvulsant medications during the perioperative period.
Learning points
In summary, we present the successful anesthetic management of an 11-year-old child with AIFM1-COXPD6. To date, there is only one previous report of anesthetic care in a similar patient, a 34-year-old woman with early childhood-onset mitochondrial encephalopathy, attributed to an AIFM1 mutation [33]. Spinal anesthesia was provided uneventfully for cesarean section. In patients with mitochondrial disorders, concern has been raised regarding the impact of both propofol and volatile anesthetic agents on mitochondrial function, prompting us to use remimazolam and remifentanil for TIVA. The clinical variability in AIFM1-COXPD6, limited patient reports, and the potential for variable response to anesthetic agents make it difficult to comment on the optimal choice of agents for intraoperative care. Additional concerns result from the end-organ involvement of these disorders which may impact CNS, upper airway, respiratory, and cardiovascular function. Additional perioperative concerns may result intraoperative handling of lactate in IV fluids and alterations in glucose metabolism. As a prolonged effect from NMBAs may be seen, intraoperative train-of-four monitoring may be indicated while sugammadex may be the optimal agent for reversal of residual neuromuscular blockade. Regardless of the agents chosen, close monitoring of postoperative cardiorespiratory function is suggested.
Acknowledgments
None to declare.
Financial Disclosure
None to declare.
Conflict of Interest
None to declare.
Informed Consent
Review of this case and presentation in this format followed the guidelines of the Institutional Review Board of Nationwide Children’s Hospital. Written consent was obtained for the use of deidentified patient information for publication.
Author Contributions
Preparation of initial, subsequent, and final drafts (ICO); direct patient care, concept, writing, and review of all drafts (JDT).
Data Availability
Any inquiries regarding supporting data availability of this study should be directed to the corresponding author.
| References | ▴Top |
- Niezgoda J, Morgan PG. Anesthetic considerations in patients with mitochondrial defects. Paediatr Anaesth. 2013;23(9):785-793.
doi pubmed - Davis RL, Liang C, Sue CM. Mitochondrial diseases. Handb Clin Neurol. 2018;147:125-141.
doi pubmed - Rahman S. Mitochondrial disease in children. J Intern Med. 2020;287(6):609-633.
doi pubmed - Filosto M, Cotti Piccinelli S, Lamperti C, Mongini T, Servidei S, Musumeci O, Tonin P, et al. Muscle pain in mitochondrial diseases: a picture from the Italian network. J Neurol. 2019;266(4):953-959.
doi pubmed - Peng Q, Ma K, Wang L, Zhu Y, Zhang Y, Rao C, Luo D, et al. Case report: a novel intronic mutation in AIFM1 associated with fatal encephalomyopathy and mitochondrial disease in infant. Front Pediatr. 2022;10:889089.
doi pubmed - Berger I, Ben-Neriah Z, Dor-Wolman T, Shaag A, Saada A, Zenvirt S, Raas-Rothschild A, et al. Early prenatal ventriculomegaly due to an AIFM1 mutation identified by linkage analysis and whole exome sequencing. Mol Genet Metab. 2011;104(4):517-520.
doi pubmed - Wischhof L, Scifo E, Ehninger D, Bano D. AIFM1 beyond cell death: An overview of this OXPHOS-inducing factor in mitochondrial diseases. EBioMedicine. 2022;83:104231.
doi pubmed - Susin SA, Lorenzo HK, Zamzami N, Marzo I, Snow BE, Brothers GM, Mangion J, et al. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature. 1999;397(6718):441-446.
doi pubmed - Hangen E, Blomgren K, Benit P, Kroemer G, Modjtahedi N. Life with or without AIF. Trends Biochem Sci. 2010;35(5):278-287.
doi pubmed - Ghezzi D, Sevrioukova I, Invernizzi F, Lamperti C, Mora M, D'Adamo P, Novara F, et al. Severe X-linked mitochondrial encephalomyopathy associated with a mutation in apoptosis-inducing factor. Am J Hum Genet. 2010;86(4):639-649.
doi pubmed - Joza N, Pospisilik JA, Hangen E, Hanada T, Modjtahedi N, Penninger JM, Kroemer G. AIF: not just an apoptosis-inducing factor. Ann N Y Acad Sci. 2009;1171:2-11.
doi pubmed - Nguyen TNA, Lai HT, Fernandes R, Dall'Olio FG, Bleriot C, Ha-Duong T, Brenner C. Apoptosis-inducing factor (AIF) at the crossroad of cell survival and cell death: implications for cancer and mitochondrial diseases. Cell Commun Signal. 2025;23(1):264.
doi pubmed - Ardissone A, Piscosquito G, Legati A, Langella T, Lamantea E, Garavaglia B, Salsano E, et al. A slowly progressive mitochondrial encephalomyopathy widens the spectrum of AIFM1 disorders. Neurology. 2015;84(21):2193-2195.
doi pubmed - Zambon AA, Ghezzi D, Baldoli C, Cutillo G, Fontana K, Sofia V, Patricelli MG, et al. Expanding the spectrum of neonatal-onset AIFM1-associated disorders. Ann Clin Transl Neurol. 2023;10(10):1844-1853.
doi pubmed - Morton SU, Prabhu SP, Lidov HGW, Shi J, Anselm I, Brownstein CA, Bainbridge MN, et al. AIFM1 mutation presenting with fatal encephalomyopathy and mitochondrial disease in an infant. Cold Spring Harb Mol Case Stud. 2017;3(2):a001560.
doi pubmed - Diodato D, Tasca G, Verrigni D, D'Amico A, Rizza T, Tozzi G, Martinelli D, et al. A novel AIFM1 mutation expands the phenotype to an infantile motor neuron disease. Eur J Hum Genet. 2016;24(3):463-466.
doi pubmed - Kettwig M, Schubach M, Zimmermann FA, Klinge L, Mayr JA, Biskup S, Sperl W, et al. From ventriculomegaly to severe muscular atrophy: expansion of the clinical spectrum related to mutations in AIFM1. Mitochondrion. 2015;21:12-18.
doi pubmed - Ellinas H, Frost EA. Mitochondrial disorders—a review of anesthetic considerations. Middle East J Anaesthesiol. 2011;21(2):235-242.
pubmed - Munlemvo DM, Syed A, Kimmet N, Hayes S, Tobias JD. Anesthetic care for a child with undiagnosed myopathic/mitochondrial disease for gastrostomy tube placement and muscle biopsy. Pediatr Anish Crit Care J. 2020;8(2):133-139.
- Yamamoto T, Miyazawa N, Yamamoto S, Kawahara H. Anesthetic management in mitochondrial encephalomyopathy: a case report. Anesth Prog. 2017;64(4):235-239.
doi pubmed - Hsieh VC, Krane EJ, Morgan PG. Mitochondrial disease and anesthesia. J Inborn Errors Metab Screen. 2017;5:1-5.
- Allison KR. Muscular dystrophy versus mitochondrial myopathy: the dilemma of the undiagnosed hypotonic child. Paediatr Anaesth. 2007;17(1):1-6.
doi pubmed - Muravchick S, Levy RJ. Clinical implications of mitochondrial dysfunction. Anesthesiology. 2006;105(4):819-837.
doi pubmed - Shipton EA, Prosser DO. Mitochondrial myopathies and anaesthesia. Eur J Anaesthesiol. 2004;21(3):173-178.
doi pubmed - Tobias JD. Clinical experience with remimazolam in pediatric anesthesiology: An educational focused review. Paediatr Anaesth. 2024;34(11):1095-1106.
doi pubmed - Yamadori Y, Yamagami Y, Matsumoto Y, Koizumi M, Nakamura A, Mizuta D, Yasuda K, et al. General anesthesia with remimazolam for a pediatric patient with MELAS and recurrent epilepsy: a case report. JA Clin Rep. 2022;8(1):75.
doi pubmed - Suzuki Y, Doi M, Nakajima Y. General anesthesia with remimazolam in a patient with mitochondrial encephalomyopathy: a case report. JA Clin Rep. 2021;7(1):51.
doi pubmed - Ueda M, Tanaka N, Momota Y, Kawaguchi M. Anesthetic management with remimazolam for adolescent mitochondrial encephalomyopathy with lactic acidosis and stroke-like episodes (MELAS): a case report. Anesth Prog. 2025;72(1):46-48.
doi pubmed - Gyurgyik N, Warren J, Miketic R, Tobias JD. Use of remimazolam as an adjunct to general anesthesia for an adolescent with MELAS syndrome. Pediatr Anish Crit Care J. 2022;10:49-55.
- Finsterer J, Stratil U, Bittner R, Sporn P. Increased sensitivity to rocuronium and atracurium in mitochondrial myopathy. Can J Anaesth. 1998;45(8):781-784.
doi pubmed - Tobias JD. Current evidence for the use of sugammadex in children. Paediatr Anaesth. 2017;27(2):118-125.
doi pubmed - Martyn JA, Richtsfeld M. Succinylcholine-induced hyperkalemia in acquired pathologic states: etiologic factors and molecular mechanisms. Anesthesiology. 2006;104(1):158-169.
doi pubmed - Anisakis P, Kalpita K, Saitic A, Valvatids D. Perioperative management of mitochondrial encephalopathy associated with a novel AIFM1 mutation in a high-risk pregnancy: a case report. Greek J Pericope Med. 2020;19:13-19.
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Medical Cases is published by Elmer Press Inc.