| Journal of Medical Cases, ISSN 1923-4155 print, 1923-4163 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Med Cases and Elmer Press Inc |
| Journal website https://jmc.elmerpub.com |
Case Report
Volume 000, Number 000, October 2025, pages 000-000

Perioperative Care of a Pediatric Patient With Beals Syndrome
Aubrey Wronaa, Jay Holladayb, Joseph D. Tobiasb, c
aHeritage College of Osteopathic Medicine - Dublin Campus, Dublin, Ohio and Ohio University, Athens, OH, USA
bDepartment of Anesthesiology & Pain Medicine, Nationwide Children’s Hospital and the Department of Anesthesiology & Pain Medicine, The Ohio State University College of Medicine, Columbus, OH, USA
cCorresponding Author: Joseph D. Tobias, Department of Anesthesiology & Pain Medicine, Nationwide Children’s Hospital, Columbus, OH 43205, USA
Manuscript submitted June 26, 2025, accepted August 28, 2025, published online October 10, 2025
Short title: Anesthesia and Beals Syndrome
doi: https://doi.org/10.14740/jmc5157
| Abstract | ▴Top |
The trismus pseudocamptodactyly syndrome (Beals syndrome) is an uncommon autosomal dominant condition first described in 1971. The disorder shares phenotypic similarities with Marfan syndrome. Affected patients classically present with two main physical features: limited excursion of the mandible and flexion deformity of the fingers that occurs with wrist extension (pseudocamptodactyly). The primary cellular defect is a mutation of the fibrillin-2 (FBN2) gene on chromosome 5q23. Mutations to this gene change the structure of the FBN2 protein, decreasing the elasticity and altering the strength of microfibrils in the connective tissue. The connective tissue defect leads to short muscle tendon units, which prevent normal growth and development. We present an 11-year-old boy with Beals syndrome who presented for anesthetic care during posterior spinal fusion (PSF). To date, there are a limited number of reports in the literature outlining anesthetic care in these patients. End-organ involvement of Beals syndrome is outlined, the potential impact on perioperative care discussed, and previous reports of anesthetic care reviewed.
Keywords: Beals syndrome; Pseudocamptodactyly; Airway management; Aortic root dilatation
| Introduction | ▴Top |
Beals syndrome (congenital contractural arachnodactyly) is an uncommon autosomal dominant condition that shares phenotypic similarities with Marfan syndrome [1-3]. Alternative nomenclatures for the condition include distal arthrogryposis type 9, trismus pseudocamptodactyly, Dutch-Kentucky or Beals-Hecht (Hecht-Beals) syndrome. The primary cellular defect is a mutation of the fibrillin-2 (FBN2) gene on chromosome 5q23. Clinical characteristics include multiple flexion contractures, arachnodactyly, severe kyphoscoliosis, abnormal pinnae and muscular hypoplasia [2]. Given its similar phenotypic presentation with Marfan syndrome, determination of its exact incidence is difficult, but estimated to be approximately 1 in 10,000. Reports have increased since the discovery of the FBN2 gene.
Affected patients classically present with multiple flexion contractures including limited excursion of the mandible and flexion deformity of the fingers that occurs with wrist extension, otherwise known as pseudocamptodactyly. Other common clinical manifestations include foot deformities, kyphoscoliosis, and short stature. Shortened muscle groups, tendons, and ligaments are responsible for limited mouth opening and joint contractures. Anatomical components of Beals syndrome, including trismus, can lead to difficulties with airway management and endotracheal intubation. Cardiac defects most commonly include interrupted aortic arch, congenital heart disease, and atrial/ventricular septal defects. Given the associated orthopedic and cardiac involvement, patients may require anesthetic care during various surgical procedures. We present an 11-year-old boy with Beals syndrome who presented for anesthetic care during posterior spinal fusion (PSF). End-organ involvement of Beals syndrome is outlined, the potential impact on perioperative care discussed, and previous reports of anesthetic care reviewed.
| Case Report | ▴Top |
Review of this case and presentation in this format followed the guidelines of the Institutional Review Board of Nationwide Children’s Hospital. The study was conducted in compliance with the ethical standards of the responsible institution on human subjects as well as with the Helsinki Declaration. The patient was an 11-year, 2-month-old, 24.2 kg male presenting for a PSF with instrumentation from T11-L3 to treat scoliosis. The diagnosis of Beals syndrome was initially confirmed during his early childhood in China, but as he was adopted, specific information was lacking. At 6 years of age, he came to the United States and the diagnosis was confirmed based on the physical exam and radiographic findings. Physical findings included a right paraspinal prominence when bending forward and a lumbar moderate left-sided paraspinal prominence with a kyphotic component. His fingers had significant clinodactyly and camptodactyly. His feet were long and slender. Along with a severe 63° kyphoscoliosis, spinal imaging showed a hypoplastic L1 vertebral body with short pedicles partnered with degenerative changes of the L1/L2 disc (Fig. 1). Other comorbid conditions included a dilated aortic root, post traumatic stress disorder, and multiple contractures of the upper and lower extremities bilaterally. An echocardiogram noted the aortic root be severely dilated with trivial aortic valve regurgitation, moderate dilation of the ascending aorta, and normal biventricular size and systolic function (Fig. 2). He had a history of two previous anesthetic events for general anesthesia during magnetic resonance (MR) imaging with reports of emergence delirium. Additionally, during the second MR under general anesthesia, neck flexion resulted in airway obstruction despite the presence of an oral airway. This led to the decision to place an air-Q size 1.5 laryngeal mask airway (LMA) which allowed neck flexion for imaging without airway obstruction. Current medications included losartan (12.5 mg twice a day) for blood pressure management related to aortic root dilatation and cetirizine (5 mg once a day) as an anti-histamine for season allergies. Preoperative physical examination revealed a Mallampati Class II airway with normal mouth opening. There was limitation of neck flexion and extension. Respiratory and cardiac examinations were unremarkable. Preoperative laboratory evaluation including coagulation function, complete blood count, electrolytes, blood urea nitrogen, and creatinine was within normal limits. The patient was held nil per os for 6 h and transported to the operating room where standard American Society of Anesthesiologists’ monitors were placed. Anesthesia was induced by the inhalation of incremental concentrations of sevoflurane in air and oxygen with the maintenance of spontaneous ventilation. A peripheral intravenous cannula (18 gauge) was placed. Once adequate bag-valve-mask ventilation was demonstrated, propofol (30 mg) and rocuronium (20 mg) were administered. The trachea was intubated with a 6.0 mm cuffed endotracheal tube (ETT) using indirect videolaryngoscopy with a Glidescope® laryngoscope. There was a grade I laryngoscope view with full view of the glottis. Following the induction of anesthesia and endotracheal intubation, a second peripheral intravenous cannula and an arterial cannula (radial artery) were placed. The patient was turned prone onto the operating room table and positioned with padding of pressure points. To facilitate neurophysiological monitoring per our usual clinical practice and intraoperative pathway, maintenance anesthesia included desflurane titrated to maintain the bispectral index at 50 - 60, a remimazolam infusion (5 - 15 µg/kg/min), methadone (1.2 mg) followed by a remifentanil infusion adjusted from 0.05 to 0.3 µg/kg/h to maintain the mean arterial pressure at 55 - 65 mm Hg, and lidocaine (1 mg/kg/h) [4]. Measures to limit intraoperative blood loss and the need for the administration of allogeneic blood included controlled hypotension, intraoperative cell salvage, and tranexamic acid (50 mg/kg bolus dosing followed by 5 mg/kg/h). Surgical site prophylaxis was provided by cefazolin (50 mg/kg) every 3 h. Dexamethasone (4 mg) and ondansetron (3 mg) were administered to prevent postoperative nausea and vomiting. There were no adverse intraoperative events. The total fluid intake was 1,500 mL including 250 mL of 5% albumin (250 mL) and 1,250 mL of Normosol®-R electrolyte solution. Intraoperative urine output was 825 mL and estimated blood loss was 50 mL. At the completion of the surgical procedure, residual neuromuscular blockade was reversed with sugammadex, the patient was turned supine, and his trachea extubated when he followed commands. Postoperative analgesia included hydromorphone delivered via a patient-controlled device, intravenous acetaminophen every 6 h, intravenous ketorolac every 6 h, and a lidocaine infusion (1 mg/kg/h for the initial 24 postoperative hours). The patient was transported to the post-anesthesia care unit and then admitted to the inpatient ward. No postoperative respiratory issues were noted. The postoperative course was uneventful, and he was discharged home on postoperative day 2.
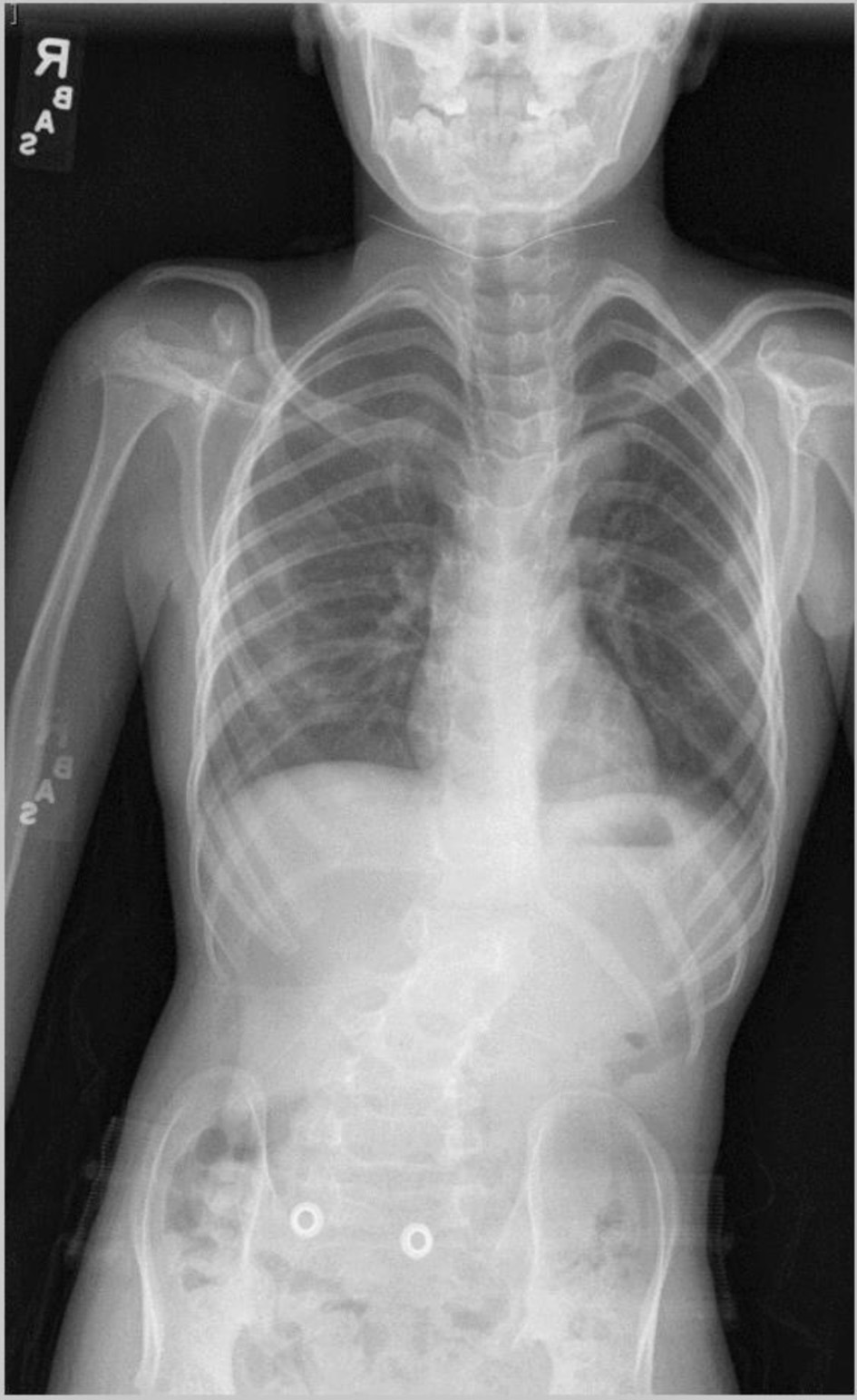 Click for large image | Figure 1. Preoperative vertebral radiograph showing scoliosis. |
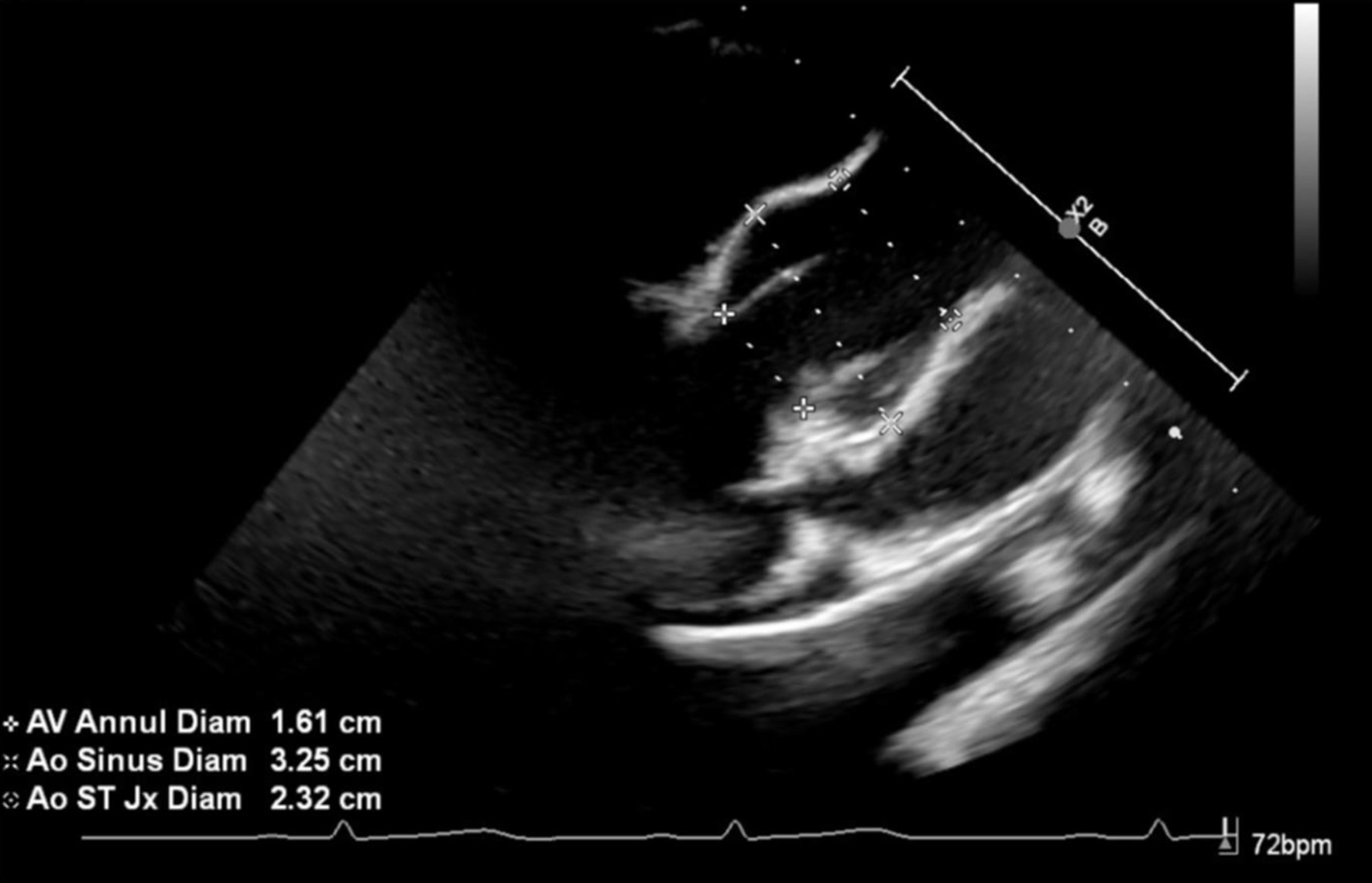 Click for large image | Figure 2. Preoperative echocardiograph image showing severe dilatation of the aortic root. |
| Discussion | ▴Top |
Beals syndrome is an autosomal dominant disorder that was first reported in 1971 [1]. A mutation of the FBN2 gene on chromosome 5q23 results in this rare connective tissue disorder. FBN2 is expressed in bone and tendons, being necessary for the control of bone growth and regulating its density. Mutations to this gene change the structure of the FBN protein, decreasing the elasticity and altering the strength of microfibrils in the connective tissue [3]. Although the clinical and phenotypic features are similar to Marfan syndrome, the incidence of cardiac abnormalities including aortic root dilatation are much lower in Beals syndrome. Additionally, the presence of multiple flexion contractures and scoliosis is characteristic of Beals syndrome. Given the multi-system involvement of Beals syndrome, anesthetic care may be required during surgical procedures to correct or palliate cardiac or orthopedic involvement. As with all anesthetic care, the first step involves a thorough preoperative history and physical examination to identify comorbid conditions related to the primary disease process with optimization of the patient’s status prior to anesthesia.
Patients with known genetic syndromes pose a variety of challenges to the anesthesia provider including the potential for difficulties with airway management, bag-valve-mask ventilation or endotracheal intubation [5, 6]. This is particularly true in Beals syndrome as the connective tissue and bone involvement can classically involve the mandible with micrognathia, trismus, and limited mouth opening. This results from fibrosis of the temporomandibular joint (TMJ) muscles, thickening and shortening of ligaments, micrognathia, and hyperplasia of the mandibular coronoid process [7-9]. Anecdotal reports, the first of which was published in 1986, have documented difficulties with airway management and endotracheal intubation (Table 1) [10-19]. Many of these have used the combination of the inhalation induction of anesthesia with the maintenance of spontaneous ventilation plus novel techniques to safely accomplish endotracheal intubation including fiberoptic guidance, use of a supraglottic airway as a conduit, or indirect videolaryngoscopy. Although inhalation induction with maintenance of spontaneous ventilation has been successful, given the airway involvement, several authors have suggested the need to have a pediatric otolaryngologist present in the operating room. In our patient, there was a previous history of airway concerns during sedation with a native airway and spontaneous ventilation for MR imaging where neck flexion resulted in airway obstruction despite the presence of an oral airway. This resulted in the need to place an LMA to overcome upper airway obstruction during neck flexion. Preoperative physical examination revealed a Mallampati Class II airway with normal mouth opening although there was limitation of neck flexion and extension. Given these concerns, endotracheal intubation was performed with indirect videolaryngoscopy using a Glidescope®. Anesthesia was induced by the inhalation of incremental concentrations of sevoflurane in air and oxygen with the maintenance of spontaneous ventilation and adequate bag-valve-mask ventilation demonstrated prior to the administration of a neuromuscular blocking agent. Additionally, the appropriate equipment for dealing with the difficult airway was readily available [6, 20].
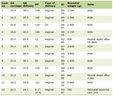 Click to view | Table 1. Previous Reports of Anesthetic Implications of Beals Syndrome |
Previous authors have outlined other specific end-organ involvement in patients with Beals syndrome that may impact perioperative care including respiratory, cardiac, and musculoskeletal involvement (Table 2). Respiratory impairment in Beals syndrome generally results from skeletal deformities (pectus excavatum or scoliosis) or rib cage abnormalities that may impact the normal pathways for lung expansion and development. As is age appropriate and feasible, preoperative pulmonary function testing may be indicated in patients with severe chest wall or vertebral deformities to assess its impact on respiratory function and plan postoperative care and monitoring.
Click to view | Table 2. End-Organ Involvement and Perioperative Concerns With Beals Syndrome |
When compared to Marfan’s syndrome, anatomical cardiac defects are less frequent and tend to be anatomically less severe in patient with Beals syndrome. Congenital cardiac defects occur in approximately 15% of patients with Beals syndrome, the most common being aortic root dilation and mitral valve prolapse, and atrial/ventricular septal defects. Less common abnormalities may include atrial/ventricular septal defects and interrupted aortic arch. Aortic root dilation tends to be milder than that seen in Marfan’s syndrome, which tends to have measurements of more than 2 standard deviations above the mean [21]. In addition to structural defects, an isolated case report noted transient cardiomyopathy with balloon-like dilation of the left ventricle [22]. Given these concerns, preoperative echocardiography is suggested.
Musculoskeletal abnormalities and contractures may impact patient positioning and vascular access. As with any comorbid condition that impacts vascular access, the use of ultrasound is suggested to facilitate peripheral venous access, arterial cannulation, and when needed, central venous access. A single case report noted a tortuous radial artery during arterial cannulation that prompted the use of an arterial cannula place in the ulnar artery, further demonstrating the utility of ultrasound.
Learning points
Genetic syndromes such as Beals syndrome may have comorbid involvement that impacts perioperative care. Individualized planning and preoperative planning can mitigate the impact of these comorbid conditions on perioperative care. Various concerns have been reported in the literature regarding perioperative care (Table 2). Of primary importance to the anesthetic provider is the potential for difficulties with direct laryngoscopy and endotracheal intubation related to trismus, bony abnormalities of the mandible, micrognathia, and limited mouth opening. Additional end-organ involvement may include restrictive lung disease related to kyphoscoliosis or pectus excavatum, and associated congenital heart defects or aortic root dilatation. Intraoperative planning should focus on a plan for airway management with maintenance of spontaneous ventilation until effective bag-valve-mask ventilation is demonstrated and the use of indirect videolaryngoscopy for endotracheal intubation.
Acknowledgments
None to declare.
Financial Disclosure
None to declare.
Conflict of Interest
None to declare.
Informed Consent
Informed consent was obtained for anesthetic care and the use of de-identified information for publication.
Author Contributions
AW: preparation of initial, subsequent, and final drafts; JDT: concept, writing, and review of all drafts; JH: perioperative care of patient, and review of final draft.
Data Availability
Any inquiries regarding supporting data availability of this study should be directed to the corresponding author.
| References | ▴Top |
- Beals RK, Hecht F. Congenital contractural arachnodactyly. A heritable disorder of connective tissue. J Bone Joint Surg Am. 1971;53(5):987-993.
pubmed - Viljoen D. Congenital contractural arachnodactyly (Beals syndrome). J Med Genet. 1994;31(8):640-643.
doi pubmed - Robinson PN, Godfrey M. The molecular genetics of Marfan syndrome and related microfibrillopathies. J Med Genet. 2000;37(1):9-25.
doi pubmed - Martin DP, Bhalla T, Thung A, Rice J, Beebe A, Samora W, Klamar J, et al. A preliminary study of volatile agents or total intravenous anesthesia for neurophysiological monitoring during posterior spinal fusion in adolescents with idiopathic scoliosis. Spine (Phila Pa 1976). 2014;39(22):E1318-1324.
doi pubmed - Butler MG, Hayes BG, Hathaway MM, Begleiter ML. Specific genetic diseases at risk for sedation/anesthesia complications. Anesth Analg. 2000;91(4):837-855.
doi pubmed - Bryant J, Krishna SG, Tobias JD. The difficult airway in pediatrics. Advan Anesth. 2013;31:31-60.
- Sreenivasan P, Peedikayil FC, Raj SV, Meundi MA. Trismus pseudocamptodactyly syndrome: a sporadic cause of trismus. Case Rep Dent. 2013;2013:187571.
doi pubmed - Beals RK. The distal arthrogryposes: a new classification of peripheral contractures. Clin Orthop Relat Res. 2005;435:203-210.
pubmed - Carlos R, Contreras E, Cabrera J. Trismus-pseudocamptodactyly syndrome (Hecht-Beals' syndrome): case report and literature review. Oral Dis. 2005;11(3):186-189.
doi pubmed - Browder FH, Lew D, Shahbazian TS. Anesthetic management of a patient with Dutch-Kentucky syndrome. Anesthesiology. 1986;65(2):218-219.
doi pubmed - Vaghadia H, Blackstock D. Anaesthetic implications of the trismus pseudocamptodactyly (Dutch-Kentucky or Hecht Beals) syndrome. Can J Anaesth. 1988;35(1):80-85.
doi pubmed - Geva D, Ezri T, Szmuk P, Gelman-Kohan Z, Shklar BZ. Anaesthesia for Hecht Beals syndrome. Paediatr Anaesth. 1997;7(2):178-179.
doi pubmed - Seavello J, Hammer GB. Tracheal intubation in a child with trismus pseudocamptodactyly (Hecht) syndrome. J Clin Anesth. 1999;11(3):254-256.
doi pubmed - Nagata O, Tateoka A, Shiro R, Kimizuka M, Hanaoka K. Anaesthetic management of two paediatric patients with Hecht-Beals syndrome. Paediatr Anaesth. 1999;9(5):444-447.
doi pubmed - Kumar A, Chandran R, Khanna P, Bhalla AP. Successful difficult airway management in a child with Hecht-Beals syndrome. Indian J Anaesth. 2012;56(6):591-592.
doi pubmed - Nasreen F, Khalid A. An infant with Beals-Hecht syndrome: An airway challenge for anaesthesiologist. Sri Lankan J Anaesthesiol. 2020;28(2):150-152.
- Vazquez-Colon CN, Lee AC. Open wide: Anesthetic management of a child with Hecht-Beals syndrome. Saudi J Anaesth. 2021;15(1):53-55.
doi pubmed - Dada RS, Hayanga JW, Abbas Khan MA, Toker A, Hayanga HK. A 36-year-old female with congenital contractural arachnodactyly and pectus excavatum requiring fourth-time redo surgical correction. Cureus. 2021;13(7):e16701.
doi pubmed - Chandramohan M, Mistry T, Balavenkatasubramanian J, Kumar Goel V, Balasubramanian S. Practical considerations in the anesthetic management of thoracolumbar scoliosis correction in a child with Hecht-Beals syndrome. Ind J Clin Anesth. 2022;9:288-290.
- Engelhardt T, Weiss M. A child with a difficult airway: what do I do next? Curr Opin Anaesthesiol. 2012;25:326-332.
- Ramos Arroyo MA, Weaver DD, Beals RK. Congenital contractural arachnodactyly. Report of four additional families and review of literature. Clin Genet. 1985;27(6):570-581.
doi pubmed - Matsumoto T, Watanabe A, Migita M, Gocho Y, Hayakawa J, Ogawa S, Shimada T, et al. Transient cardiomyopathy in a patient with congenital contractural arachnodactyly (Beals syndrome). J Nippon Med Sch. 2006;73(5):285-288.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Medical Cases is published by Elmer Press Inc.