| Journal of Medical Cases, ISSN 1923-4155 print, 1923-4163 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Med Cases and Elmer Press Inc |
| Journal website https://jmc.elmerpub.com |
Case Report
Volume 000, Number 000, August 2025, pages 000-000

Thinking Outside the Box: Case Report of a Rare Quadricuspid Aortic Valve as an Underrecognized Cause of Heart Failure and Atrial Fibrillation
Klevis Mihalia, b, d, Birgit Markusa, Bernhard Schieffera, Marcus Bauerb, c, Julian Kreutza, c
aDepartment of Cardiology, Angiology, and Intensive Care Medicine, Philipps-Universitat Marburg, Germany
bDepartment of Cardiology, St. Vincenz-Krankenhaus Datteln, Germany
cThese authors contributed equally to this work.
dCorresponding Author: Klevis Mihali, Department of Cardiology, Angiology, and Intensive Care Medicine, Philipps-Universitat Marburg, 35043 Marburg, Germany
Manuscript submitted June 14, 2025, accepted July 18, 2025, published online August 7, 2025
Short title: Rare Quadricuspid Aortic Valve
doi: https://doi.org/10.14740/jmc5153
| Abstract | ▴Top |
Quadricuspid aortic valve (QAV) is a rare congenital anomaly with an estimated incidence of 0.008% to 0.043% based on autopsy and echocardiographic studies. Although often asymptomatic, it can lead to progressive aortic regurgitation (AR), left ventricular (LV) dysfunction, and arrhythmias such as atrial fibrillation (AF). Due to its rarity, QAV is often misdiagnosed or discovered incidentally, highlighting the need for advanced cardiac imaging in young patients presenting with unexplained heart failure symptoms and arrhythmias. We present the case of a 41-year-old female patient who was admitted with new-onset dyspnea classified as New York Heart Association (NYHA) class III and palpitations due to persistent AF with a European Heart Rhythm Association (EHRA) symptom class 2b. There was no family history of congenital or structural heart disease, with arterial hypertension being the only identified predisposing condition. Initial transthoracic echocardiography revealed moderate AR, but more detailed transesophageal echocardiography performed before pulmonary vein isolation incidentally revealed a QAV. Further cardiac magnetic resonance imaging confirmed normal aortic root dimensions with early LV remodeling. The patient was managed conservatively with rate control, anticoagulation, and regular follow-up to monitor disease progression. This case highlights the importance of advanced imaging techniques in the diagnosis of rare structural heart abnormalities in young patients presenting with unexplained heart failure symptoms and arrhythmias. Early identification of QAV allows for timely medical intervention, optimal patient monitoring, and prevention of long-term complications. Regular follow-up is essential to monitor disease progression and determine the need for surgical intervention.
Keywords: Quadricuspid aortic valve; Congenital heart disease; Aortic valve dysfunction; Aortic regurgitation; Atrial fibrillation
| Introduction | ▴Top |
Quadricuspid aortic valve (QAV) is a rare congenital malformation with an estimated prevalence between 0.008% and 0.043% [1]. Historically diagnosed post-mortem or during valve surgery, advances in echocardiography and cardiac imaging have significantly improved detection rates [2]. Despite its rarity, QAV is clinically significant due to its frequent association with aortic regurgitation (AR), left ventricular (LV) remodeling and in some cases, arrhythmias such as atrial fibrillation (AF). Over time, these structural abnormalities can result in hemodynamic disturbances, LV volume overload, and ultimately heart failure. In addition, QAV may be associated with other congenital anomalies, including coronary artery malformations, hypertrophic cardiomyopathy, subaortic stenosis, patent ductus arteriosus, ascending aorta coarctation, and ventricular septal defects [3-5]. The embryological origins of QAV remain uncertain, but proposed mechanisms include abnormal septation of the conotruncus, defective mesenchymal proliferation, or incomplete division of the valve cusps [6]. The Hurwitz and Roberts classification system describes seven morphological subtypes, of which type B (three larger and one smaller cusp) is the most common [7]. The classification ranges from type A (four equal cusps) to type G, with variations based on the relative size and symmetry of the cusps. Type C, as observed in our patient, consists of two larger and two smaller cusps. These morphological differences are clinically relevant, as asymmetric cusp configurations, especially in types B through G, can lead to unequal leaflet stress, impaired coaptation, and a higher likelihood of developing aortic regurgitation over time. Clinically, many patients with QAV remain asymptomatic for years, with symptoms often occurring in middle adulthood due to progressive valvular dysfunction [8]. The primary pathological consequence of QAV is AR, which can often require valve replacement in the fifth or sixth decade of life if it progresses [1]. While transthoracic echocardiography (TTE) is the initial diagnostic tool, transesophageal echocardiography (TEE) provides superior visualization, and cardiac magnetic resonance (CMR) imaging or computed tomography angiography (CTA) may further aid in assessment. Management of QAV varies from observation in asymptomatic cases to surgical intervention in those with severe regurgitation or LV dysfunction. Early detection and regular follow-ups are crucial for optimizing patient outcomes, especially given its potential for progressive deterioration.
| Case Report | ▴Top |
A 41-year-old female patient presented to the emergency department with new-onset dyspnea in New York Heart Association (NYHA) class III and palpitations attributed to AF with a European Heart Rhythm Association (EHRA) score of 2b. She denied chest pain, syncope, alcohol consumption, or tobacco use and reported no symptoms suggestive of obstructive sleep apnea. Her medical history was notable only for arterial hypertension and her body mass index was 24.3 kg/m2. There was no known family history of congenital or structural heart disease.
Physical examination revealed an arrhythmic heart rate of 110 beats per minute. Auscultation identified a grade II/VI high-pitched, early diastolic decrescendo murmur at the left third-fourth intercostal space along the sternal border. A resting electrocardiogram showed AF with a rapid ventricular response. Laboratory tests showed an N-terminal pro-B-type natriuretic peptide (NT-proBNP) level of 780 pg/mL and normal levels of troponin and thyroid hormones. TTE demonstrated moderate AR with a pressure half-time of 390 ms and a preserved LV ejection fraction (LVEF) of 55%, without evidence of LV dilation. Given the patient’s young age, the presence of AF, and unexplained symptoms in the absence of severe AR, further cardiovascular imaging was performed. TEE confirmed the presence of a QAV with Hurwitz and Roberts type C morphology and moderate AR based on an integrative semi-quantitative assessment, including a vena contracta of 0.31 cm and a pressure half-time of 390 ms (Fig. 1). Due to AF and suboptimal conditions for volumetric assessment, a comprehensive quantitative evaluation could not be reliably performed. CMR imaging showed normal aortic root dimensions, no evidence of significant aortopathy, and preserved LV function with early remodeling changes (Fig. 2). The severity of AR was classified as moderate in the CMR report; however, no quantitative parameters such as regurgitant volume or regurgitant fraction were documented. The presence of symptoms such as exertional dyspnea and persistent AF, together with elevated NT-proBNP values and an H2FPEF score of 6 indicated a high probability of heart failure with preserved ejection fraction (HFpEF) being the underlying disease. The patient was initiated on guideline-directed medical therapy for HFpEF and anticoagulated with apixaban (CHA2DS2-VASc score of 3) to reduce the risk of thromboembolic events. Despite medical management, she continued to experience episodes of AF. Given her young age, recent-onset AF, and the presence of moderate AR, rhythm control was preferred over rate control to preserve atrial contraction and optimize diastolic filling [9]. Therefore, pulmonary vein isolation via catheter ablation was performed successfully.
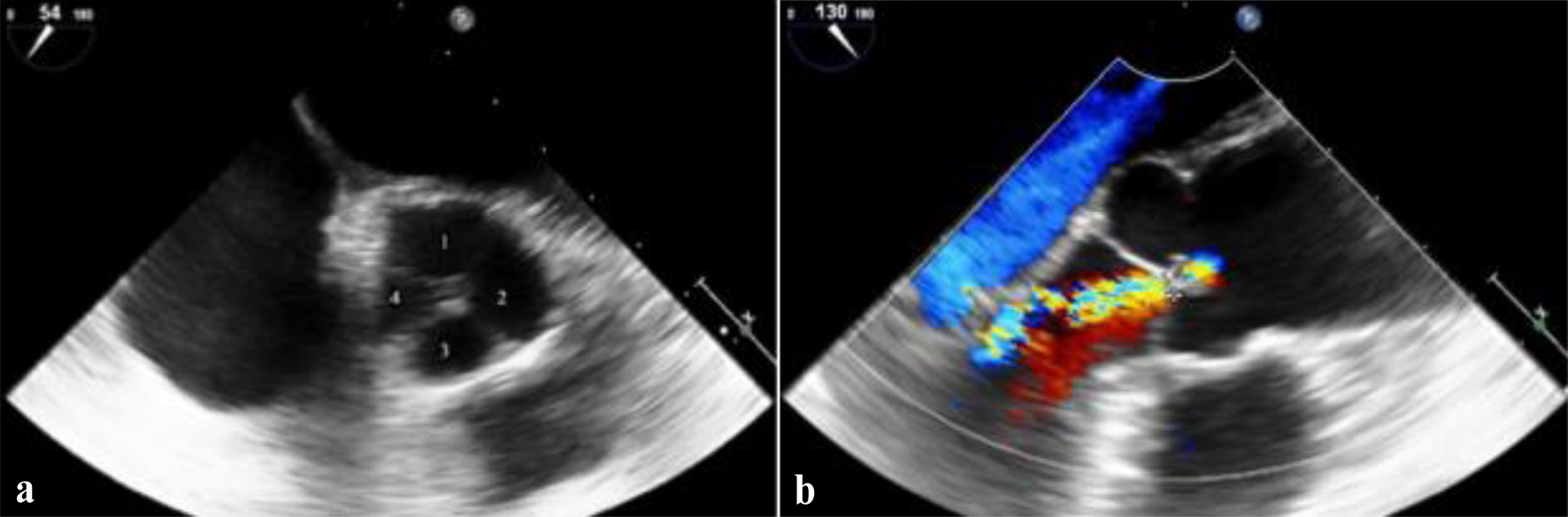 Click for large image | Figure 1. Transesophageal echocardiography views of the aortic valve. (a) Mid-esophageal short-axis view at 54°, demonstrating a quadricuspid aortic valve (Hurwitz and Roberts type C) with four distinct cusps. The individual cusps are labeled to aid visualization. (b) Mid-esophageal long-axis view at 130° with color Doppler, showing central aortic regurgitation jet consistent with moderate aortic insufficiency. |
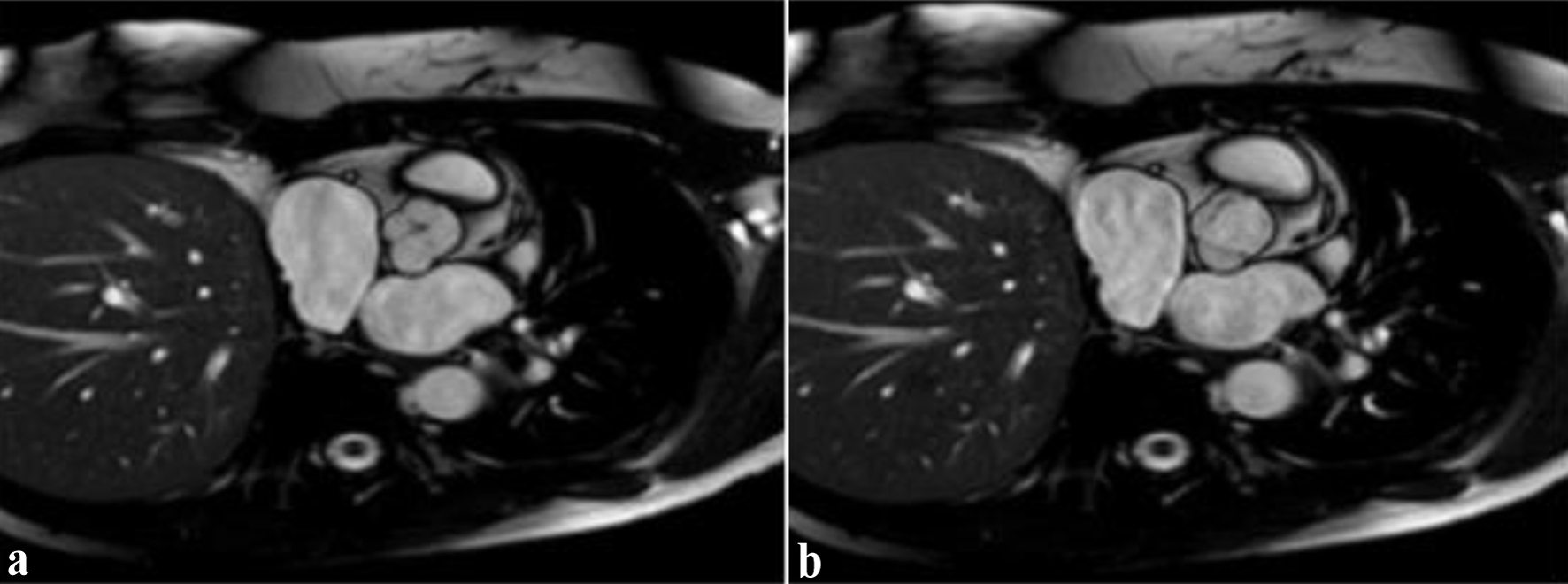 Click for large image | Figure 2. Cardiac magnetic resonance imaging in short-axis view demonstrating quadricuspid aortic valve morphology. (a) Diastolic phase: incomplete cusp coaptation with central aortic regurgitation orifice, consistent with moderate aortic regurgitation. (b) Systolic phase: the aortic valve leaflets are fully open, visualizing four distinct cusps. |
As the patient remained asymptomatic from a valvular standpoint, a conservative approach was adopted with close cardiological follow-up and serial echocardiographic monitoring. Given the diagnosis of QAV and its potential for progressive AR, we established a structured follow-up strategy with echocardiographic evaluations every 6 months and provided patient counseling on infective endocarditis precautions. Future surgical intervention may be considered in the event of progressive AR or LV dysfunction.
| Discussion | ▴Top |
This case underscores the importance of a thorough cardiovascular workup in young patients presenting with unexplained heart failure symptoms and arrhythmias. Although bicuspid aortic valve remains the most common congenital anomaly of the aortic valve, rarer entities such as QAV should also be considered, particularly in cases of unexplained AR [10]. In such scenarios, reliance on TTE alone may lead to underdiagnosis. Therefore, advanced imaging modalities like TEE or CMR should be employed to enhance diagnostic accuracy and detect subtle congenital abnormalities.
Our case highlights the diagnostic overlap between valvular heart disease and HFpEF. QAV is often associated with AR due to impaired cusp coaptation leading to progressive LV volume overload and remodeling. Prolonged volume stress may also result in left atrial dilation, predisposing to arrhythmias such as AF, even in the absence of overt ventricular dysfunction. While AF is a common arrhythmia in valvular heart disease, its presence in a young patient with no known structural heart disease warrants further imaging to rule out congenital anomalies such as QAV. A major challenge with QAV is its frequent under-diagnosis on routine echocardiography. TTE, although widely used, has limited sensitivity for detecting QAV. To date, no large-scale studies have reported specific sensitivity values for TTE or TEE in diagnosing QAV, likely due to the rarity of the condition. Most available data stem from case reports or small series, which describe TTE as having limited sensitivity, especially when short-axis imaging is suboptimal [11]. TEE is consistently regarded as superior to TTE for visualizing cusp morphology and remains the preferred modality for definitive diagnosis [2]. Although CMR offers high spatial resolution and is non-invasive, it may not consistently provide the same level of detail regarding cusp structure. Therefore, the two modalities should be considered complementary. In addition, CMR and CTA play a crucial role in the assessment of LV function, aortic root dimensions, and associated cardiovascular abnormalities, particularly when surgical planning is required. Management of QAV depends on the severity of AR and associated symptoms. Asymptomatic patients with mild to moderate AR can be managed conservatively with regular echocardiographic follow-up to monitor progression. In cases of progressive LV dilatation or worsening LV function, surgical intervention should be considered. Although specific long-term data in QAV are limited, most patients with QAV-related AR progress to require intervention (66.7% in small series), usually between the fifth and sixth decades [12, 13]. Five- and 10-year survival rates after surgery are excellent (about 90% and 85%, respectively) [14]. Current guidelines recommend valve surgery for symptomatic patients with severe AR, or for asymptomatic patients with LVEF ≤ 50%, LV end-systolic diameter (LVESD) > 50 mm, or indexed LVESD > 25 mm/m2 [15]. Valve replacement remains the most common treatment; however, valve-sparing procedures and transcatheter aortic valve implantation have been explored in selected cases [16-19]. While QAV does not inherently increase the risk of infective endocarditis, abnormal cusp coaptation can create turbulent flow that could theoretically favor bacterial colonization [20]. However, guidelines do not recommend routine antibiotic prophylaxis in these patients [21].
Beyond structural and hemodynamic considerations, accumulating evidence points to the role of immune cell activation and inflammation in the progression of cardiovascular disease. In both congenital and acquired valvular disorders, immune mechanisms, such as monocyte infiltration, T-cell subpopulation dynamics, and the interplay between innate and adaptive immunity, may contribute to valvular degeneration and myocardial remodeling [22-24]. While such processes have not been specifically studied in QAV, they may represent an underrecognized aspect of disease progression and offer a valuable direction for future research.
Conclusion
Early identification of rare congenital anomalies such as QAV is crucial in young patients presenting with unexplained heart failure symptoms and arrhythmias. Multimodal cardiac imaging, combined with structured diagnostic tools such as the H2FPEF score, can uncover overlapping etiologies such as moderate AR and HFpEF. Conservative management may be appropriate in stable patients, but close follow-up is essential to monitor for disease progression. Delayed or missed diagnosis may lead to progressive valvular dysfunction and irreversible LV remodeling, ultimately necessitating earlier surgical intervention. Further research is warranted to better define the long-term outcomes of valve-sparing and minimally invasive interventions in patients with QAV.
Acknowledgments
None to declare.
Financial Disclosure
This research received no specific grant from any funding agency.
Conflict of Interest
The authors declare that they have no conflict of interest.
Informed Consent
Written informed consent was obtained from the patient for the publication of this case report.
Author Contributions
Conceptualization: Klevis Mihali and Marcus Bauer; writing manuscript: Klevis Mihali and Julian Kreutz; writing review and editing: Birgit Markus, Bernhard Schieffer, and Marcus Bauer; visualization: Klevis Mihali and Marcus Bauer; supervision: Julian Kreutz, Birgit Markus, and Bernhard Schieffer. All authors have read and agreed to the published version of the manuscript.
Data Availability
The authors declare that all data generated or analyzed during this case are included in this article.
| References | ▴Top |
- Hurwitz LE, Roberts WC. Quadricuspid semilunar valve. Am J Cardiol. 1973;31(5):623-626.
doi pubmed - Saith S, Saith S, Murthy A. Quadricuspid aortic valve: an introduction for clinicians. Cardiol Res. 2022;13(1):2-10.
doi pubmed - Seol SH, Kim U, Cho HJ, Kim DK, Kim DI, Kim DS. Quadricuspid aortic valve with patent ductus arteriosus. Tex Heart Inst J. 2010;37(6):726-727.
pubmed - Demirkol S, Balta S, Arslan Z, Unlu M, Kucuk U, Iyisoy A. Association of quadricuspid aortic valve and ventricular septal defect in a patient who had undergone atrial septal defect surgery. Kardiol Pol. 2013;71(5):546.
doi pubmed - Holt NF, Sivarajan M, Mandapati D, Printsev Y, Elefteriades JA. Quadricuspid aortic valve with aortic insufficiency: case report and review of the literature. J Card Surg. 2007;22(3):235-237.
doi pubmed - Gulyasy B, Lopez-Candales A, Reis SE, Levitsky S. Quadricuspid aortic valve: an unusual echocardiographic finding and a review of the literature. Int J Cardiol. 2009;132(2):e68-71.
doi pubmed - Timperley J, Milner R, Marshall AJ, Gilbert TJ. Quadricuspid aortic valves. Clin Cardiol. 2002;25(12):548-552.
doi pubmed - Yuan SM. Quadricuspid aortic valve: a comprehensive review. Braz J Cardiovasc Surg. 2016;31(6):454-460.
doi pubmed - Kirchhof P, Camm AJ, Goette A, Brandes A, Eckardt L, Elvan A, Fetsch T, et al. Early rhythm-control therapy in patients with atrial fibrillation. N Engl J Med. 2020;383(14):1305-1316.
doi pubmed - Roberts WC. The congenitally bicuspid aortic valve. A study of 85 autopsy cases. Am J Cardiol. 1970;26(1):72-83.
doi pubmed - Savino K, Quintavalle E, Ambrosio G. Quadricuspid aortic valve: a case report and review of the literature. J Cardiovasc Echogr. 2015;25(3):72-76.
doi pubmed - Tutarel O. The quadricuspid aortic valve: a comprehensive review. J Heart Valve Dis. 2004;13(4):534-537.
pubmed - Janssens U, Klues HG, Hanrath P. Congenital quadricuspid aortic valve anomaly associated with hypertrophic non-obstructive cardiomyopathy: a case report and review of the literature. Heart. 1997;78(1):83-87.
doi pubmed - Tsang MY, Abudiab MM, Ammash NM, Naqvi TZ, Edwards WD, Nkomo VT, Pellikka PA. Quadricuspid aortic valve: characteristics, associated structural cardiovascular abnormalities, and clinical outcomes. Circulation. 2016;133(3):312-319.
doi pubmed - Otto CM, Nishimura RA, Bonow RO, Carabello BA, Erwin JP, 3rd, Gentile F, Jneid H, et al. 2020 ACC/AHA guideline for the management of patients with valvular heart disease: executive summary: a report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation. 2021;143(5):e35-e71.
doi pubmed - Schmidt KI, Jeserich M, Aicher D, Schafers HJ. Tricuspidization of the quadricuspid aortic valve. Ann Thorac Surg. 2008;85(3):1087-1089.
doi pubmed - Liu Y, Zhai M, Mao Y, Xu C, Ma Y, Li L, Jin P, et al. Transcatheter aortic valve replacement in patients with quadricuspid aortic valve in a single center. Front Cardiovasc Med. 2022;9:1011466.
doi pubmed - Nishimura RA, Otto CM, Bonow RO, Carabello BA, Erwin JP, 3rd, Guyton RA, O'Gara PT, et al. 2014 AHA/ACC guideline for the management of patients with valvular heart disease: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation. 2014;129(23):e521-643.
doi pubmed - Jagannath AD, Johri AM, Liberthson R, Larobina M, Passeri J, Tighe D, Agnihotri AK. Quadricuspid aortic valve: a report of 12 cases and a review of the literature. Echocardiography. 2011;28(9):1035-1040.
doi pubmed - Takeda N, Ohtaki E, Kasegawa H, Tobaru T, Sumiyoshi T. Infective endocarditis associated with quadricuspid aortic valve. Jpn Heart J. 2003;44(3):441-445.
doi pubmed - Wilson W, Taubert KA, Gewitz M, Lockhart PB, Baddour LM, Levison M, Bolger A, et al. Prevention of infective endocarditis: guidelines from the American Heart Association: a guideline from the American Heart Association Rheumatic Fever, Endocarditis and Kawasaki Disease Committee, Council on Cardiovascular Disease in the Young, and the Council on Clinical Cardiology, Council on Cardiovascular Surgery and Anesthesia, and the Quality of Care and Outcomes Research Interdisciplinary Working Group. J Am Dent Assoc. 2008;139(Suppl):3S-24S.
doi pubmed - Kumar V, Prabhu SD, Bansal SS. CD4(+) T-lymphocytes exhibit biphasic kinetics post-myocardial infarction. Front Cardiovasc Med. 2022;9:992653.
doi pubmed - Rurik JG, Aghajanian H, Epstein JA. Immune cells and immunotherapy for cardiac injury and repair. Circ Res. 2021;128(11):1766-1779.
doi pubmed - Abplanalp WT, John D, Cremer S, Assmus B, Dorsheimer L, Hoffmann J, Becker-Pergola G, et al. Single-cell RNA-sequencing reveals profound changes in circulating immune cells in patients with heart failure. Cardiovasc Res. 2021;117(2):484-494.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Medical Cases is published by Elmer Press Inc.