| Journal of Medical Cases, ISSN 1923-4155 print, 1923-4163 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Med Cases and Elmer Press Inc |
| Journal website https://jmc.elmerpub.com |
Case Report
Volume 16, Number 7, July 2025, pages 259-266

Secondary Central Nervous System Lymphoma Involving Meninges: A Rare Case Report and a Comprehensive Review of Peripheral T-Cell Lymphoma, Not Otherwise Specified
Mrudula Thiriveedia, Muralidhar Idamakantib, f ![]() , Siddharth
Patela, Rafik ElBeblawya, Sujatha Baddamc, Bala
Nimmanac, Virginia Daileyd, Rishi Patele
, Siddharth
Patela, Rafik ElBeblawya, Sujatha Baddamc, Bala
Nimmanac, Virginia Daileyd, Rishi Patele
aDepartment of Internal Medicine, Decatur Morgan Hospital, Decatur, AL 35601,
USA
bAdult Inpatient Medical Services (AIMS), Presbyterian Healthcare Services
(PHS), Albuquerque, NM 87106, USA
cDepartment of Internal Medicine, Huntsville
Hospital, Huntsville, AL 35801, USA
dDepartment of Pathology, Decatur Morgan
Hospital, Decatur, AL 35601, USA
eDepartment of Hematology Oncology, Decatur
Morgan Hospital, Decatur, AL 35601, USA
fCorresponding Author: Muralidhar
Idamakanti, bAdult Inpatient Medical Services (AIMS), Presbyterian Healthcare Services (PHS),
Albuquerque, NM 87106, USA
Manuscript submitted May 29, 2025, accepted July 12, 2025, published online July 19,
2025
Short title: Secondary CNS Lymphoma Involving Meninges
doi:
https://doi.org/10.14740/jmc5149
| Abstract | ▴Top |
Peripheral T-cell lymphoma, not otherwise specified (PTCL-NOS), is a rare and aggressive subtype of non-Hodgkin lymphoma (NHL) that arises from mature T or natural killer (NK) cells, accounting for about 5% of all NHL cases. While PTCL-NOS typically involves lymph nodes, extranodal sites such as the skin, gastrointestinal tract, liver, and lungs can also be affected. Central nervous system (CNS) involvement is extremely rare, especially at the time of initial presentation. When it does occur, the brain is most commonly affected, followed by the spinal cord and meninges. We present a rare case of PTCL-NOS with secondary CNS lymphoma involving the meninges at initial diagnosis. Our patient is a 75-year-old male with multiple comorbidities who presented with several weeks of intermittent headaches. Imaging showed multiple extra-axial brain lesions with infiltration into extracranial soft tissues, epidural space, meninges, and brain parenchyma. A subsequent lymph node biopsy confirmed PTCL-NOS. He was started on rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone (R-CHOP), with plans for high-dose methotrexate. Thereafter, the patient was readmitted several times due to various complications and expired approximately 2.5 months after diagnosis. We conclude that secondary CNS involvement in PTCL-NOS is very rare and has a poor prognosis, with a median survival after CNS diagnosis of about 1.1 months. Early diagnosis and tailored treatment strategies, including CNS-penetrating agents, are essential. Continued research is needed to better understand and improve outcomes for this aggressive disease.
Keywords: Lymphoma; Non-Hodgkin lymphomas; Peripheral T-cell lymphoma; Peripheral T-cell lymphoma, not otherwise specified; Primary CNS lymphoma; Secondary CNS lymphoma; Methotrexate
| Introduction | ▴Top |
Peripheral T-cell lymphoma, not otherwise specified (PTCL-NOS), is a heterogeneous group of predominantly nodal T-cell lymphomas that account for 30-40% of peripheral T-cell lymphoma (PTCL) cases and about 5% of all non-Hodgkin lymphoma (NHL) cases [1-3]. A common presentation of PTCL-NOS is lymphadenopathy with or without extranodal involvement. Commonly involved sites are the skin, liver, gastrointestinal tract, salivary glands, and lungs. Involvement of the central nervous system (CNS) is rare and typically signals a poor prognosis.
PTCL accounts for only 2% of all primary CNS lymphoma (PCNSL) cases and 2-6% of all secondary CNS lymphoma (SCNSL) cases [4-7]. CNS involvement in PTCL-NOS is not common compared to other types of PTCL and NHL [4, 5, 8]. Five-year overall survival for all kinds of PTCL-NOS is 30-35% [9-11]. The median survival time for PTCL-NOS after CNS involvement is approximately 1.1 months [8, 12]. Early diagnosis and treatment are imperative for improved clinical outcomes. Here, we present a rare case of PTCL-NOS with SCNSL with meningeal involvement at the time of initial diagnosis.
| Case Report | ▴Top |
A 75-year-old Caucasian male with a history of essential hypertension, type 2 diabetes mellitus dependent on insulin, chronic obstructive pulmonary disease, hyperlipidemia, gastroesophageal reflux disease, chronic alcohol abuse with half a pint of vodka for many years, and a 50 pack-year smoking history presented with intermittent headaches ongoing for several weeks. The patient denies any chronic history of headaches before this presentation. The headaches occurred in the bifrontal to temporal area, were often pulsatile, occasionally radiated to the occipital areas, and worsened at night. He denied experiencing visual disturbances, weakness, numbness, neck stiffness, fever, or systemic symptoms. Vital signs remained stable. Upon examination, he was alert and oriented to name, place, time, and situation. Palpable, non-tender masses were noted over both temporal regions. Neurological examination showed intact muscle strength and sensation in both upper and lower extremities, grossly intact cranial nerves II-XII, with normal speech and comprehension. The general physical examination was unremarkable. Laboratory tests upon admission significant for elevated glucose (210 mg/dL) and mildly elevated creatinine (1.3 mg/dL).
Initial computed tomography (CT) of the head revealed multiple extra-axial, dural-based lesions with calvarial extensions and with involvement of meninges, epidural space, and bilateral temporal, parietal, and right frontal lobes (Fig. 1). Magnetic resonance imaging (MRI) confirmed a similar infiltrative process extending into the epidural space, meninges and brain parenchymal lesions, and associated vasogenic edema causing extrinsic compression of the right frontal lobe (Fig. 2a, b). The patient was started on dexamethasone 4 mg every 6 h, which resulted in symptomatic improvement. CT scans of the chest, abdomen, and pelvis were performed to search for any primary tumor. The CT scans revealed enhancing soft tissue masses in the right lateral supraclavicular space, partially extending into the neck, along with metastatic mediastinal and left axillary nodes, and no other evidence of malignancy or metastatic disease.
 Click for large image |
Figure 1. Initial non-contrast computed tomography (CT) scan of the head demonstrating bilateral extra-axial dural-based masses (red arrows). |
 Click for large image |
Figure 2. MRI of the brain with contrast showing an infiltrative process extending from the extracranial soft tissues into the epidural space bilaterally (red arrows), with associated brain parenchymal lesion, and vasogenic edema in the right frontal lobe causing extrinsic compression (white arrow) (a) and diffuse meningeal enhancement (white arrows) (b). MRI: magnetic resonance imaging. |
A positron emission tomography (PET) scan revealed widespread fluorodeoxyglucose (FDG)-avid lesions involving the right supraclavicular and left axillary lymph nodes (Fig. 3). Initial attempts at lumbar puncture were unsuccessful, and further attempts were aborted due to increased risks compared to benefits, as per the neurologist. An excisional biopsy of a left axillary lymph node identified PTCL-NOS (Fig. 4a, b). Immunohistochemical staining was crucial for diagnosis and demonstrated strong positivity for CD3, CD4, CD43, and B-cell lymphoma 2 (BCL2), along with scattered staining of CD15, weak positivity for CD30 and CD45, and a Ki-67 proliferation rate exceeding 90% (Figs. 5, 6). The brain biopsy was deferred due to the presumed higher risk of complications from the procedure and the availability of a confirmatory diagnosis from the lymph node biopsy. The patient was diagnosed with Lugano stage IV disease [11]. He received one cycle of rituximab, cyclophosphamide, vincristine, and prednisone (R-CHOP) after preliminary pathology results showed high-grade lymphoma with a high Ki-67 rate. With the final biopsy results, the plan was to escalate to high-dose methotrexate (HD-MTX) for CNS penetration at a tertiary facility to measure methotrexate levels precisely. A post-chemotherapy MRI of the brain indicated marked improvement (Fig. 7). Brentuximab vedotin, cyclophosphamide, doxorubicin, and prednisone (BV-CHP) were not administered due to weak positivity of CD30 and insufficient data regarding its efficiency in CNS disease.
 Click for large image |
Figure 3. PET scan showing FDG-avid lesions in the right supraclavicular and left axillary lymph node regions (arrows). PET: positron emission tomography; FDG: fluorodeoxyglucose. |
 Click for large image |
Figure 4. Hematoxylin and eosin (H&E) stained section of the lymph node biopsy showing diffuse effacement of nodal architecture by a dense infiltrate of monomorphic lymphoid tumor cells (white arrows) (a) with prominent arborized endothelial venules (red arrow) (b). |
 Click for large image |
Figure 5. Immunohistochemistry demonstrating strong and diffuse membranous CD4 staining across the neoplastic lymphoid population, consistent with a T-helper cell phenotype. |
 Click for large image |
Figure 6. Immunohistochemistry demonstrating neoplastic lymphoid population with sparse scattered CD15 staining (blue arrows). |
 Click for large image |
Figure 7. Repeat MRI of the brain with contrast demonstrating marked improvement in the previously noted irregular dural thickening (red arrows) and significant reduction of the mass-like thickening anterior to the right frontal lobe (white arrow) following initial treatment. MRI: magnetic resonance imaging. |
Unfortunately, the patient required multiple admissions thereafter due to sepsis, gastrointestinal bleeding, acute kidney injury, and atrial fibrillation with rapid ventricular response. During his last admission, he underwent an upper endoscopy that revealed no signs of gastrointestinal lymphoma but indicated duodenitis with active bleeding that required cauterization. He was stabilized with supportive care and diligent management of other comorbidities and was discharged for inpatient rehabilitation. Overall performance rapidly declined, and the patient could not tolerate further therapy. He ultimately passed away approximately 2.5 months after diagnosis.
| Discussion | ▴Top |
Peripheral T-cell lymphomas (PTCLs) are a diverse and relatively rare group of NHL that originate from mature T cells and natural killer (NK) cells. PTCLs account for 10-15% of NHL cases, and their prevalence varies based on geographic and demographic factors. PTCLs are more common in Asian countries than in the Western world. PTCLs are divided into several subtypes based on the clinical, pathological, and molecular characteristics and are categorized using the World Health Organization (WHO) fifth edition or the International Consensus Classification systems [1-3] (Table 1).
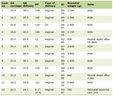 Click to view |
Table 1. Common Subtypes of Peripheral T-Cell
Lymphomas in the Order of Frequency of Occurrence |
PTCL-NOS is a heterogeneous and predominantly nodal subtype, and it is the most common form of PTCL. It accounts for 30-40% of all PTCL cases and approximately 5% of all NHL cases [1-3]. It is a diagnosis of exclusion, which is classified when a T-cell lymphoma does not fit into any other well-defined PTCL category. PTCL-NOS primarily affects older adults, with a median age at diagnosis of 60 years [10, 13]. The peak incidence of PTCL-NOS occurs among individuals aged 75 to 80 years. There is a male predominance, with a male-to-female ratio of approximately 1.9:1 [10, 13]. A significant proportion of patients present with advanced disease (Ann Arbor stages III and IV) at the time of diagnosis. In a study of 340 PTCL-NOS patients, 69% had advanced-stage disease [10]. These epidemiological characteristics highlight the aggressive nature of PTCL-NOS and the importance of early detection and treatment.
The pathogenesis of PTCL is poorly understood due to its heterogeneity. However, it is believed to involve genetic and epigenetic alterations that affect T-cell signaling, immune evasion, and apoptotic pathways. Common genetic abnormalities in PTCL include TET2, DNMT3A, and IDH2 mutations (affecting DNA methylation and gene expression), RHOA mutations (involved in cytoskeletal dynamics and signaling), and alterations in the JAK/STAT pathway (which promote tumor growth and survival) [14-16]. Recent molecular studies have identified two distinct genetic subtypes within PTCL-NOS, primarily based on gene expression profiles that include GATA3 and TBX21 overexpression. Patients with the TBX21 subtype generally have a better prognosis than those with the GATA3 subtype [14-16]. Immunophenotypically, PTCL-NOS expresses pan-T cell markers such as CD2, CD3, CD4, and CD8, with variable expression of CD30 and CD15, and downregulation of CD5 and CD7. The most common marker in nodal disease is CD4. CD15 is associated with adverse outcomes in PTCL-NOS [10, 15]. The Ki-67 proliferation index for PTCL-NOS is often high, reflecting the aggressive nature of the disease [14, 15].
PTCL-NOS is an aggressive disease with a diverse clinical presentation. Common clinical characteristics include B symptoms presenting in 35% of cases (fever, night sweats, fatigue, and unexplained weight loss), lymphadenopathy in 35-40% of cases, hepatosplenomegaly in roughly 20% of cases, skin lesions (rashes, nodules, or plaques in some cases) and gastrointestinal manifestations [3, 10, 13]. Approximately 50% of cases have both nodal and extranodal involvement, and 10-15% of cases have exclusive extranodal disease. Extranodal disease is commonly seen in the skin and gastrointestinal tract. Pulmonary and salivary gland involvement is less common, and involvement of the CNS is rare [3, 10, 13]. Bone marrow involvement with associated cytopenias is seen in around 20% of cases [17]. The International Prognostic Index (IPI) is commonly used to prognosticate PTCL-NOS. Risk factors at diagnosis that indicate poor prognosis include age older than 60 years, Ann Arbor stage III or IV, elevated serum lactate dehydrogenase (LDH), poor performance status, and more than one site of extranodal involvement [10, 11] (Table 2).
Click to view |
Table 2. Clinical Manifestations of
PTCL-NOS |
CNS disease in PTCL is uncommon and is even rarer in PTCL-NOS, with reported rates varying across studies. Adult T-cell leukemia/lymphoma has the highest incidence of CNS disease among all subtypes of PTCL [4-7]. PCNSL refers to lymphoma that originates within the CNS without evidence of systemic disease at the time of diagnosis, while SCNSL refers to systemic lymphoma with CNS spread, either at initial presentation or as a relapse during or after treatment [4, 8, 18-20]. PTCL accounts for only 2% of all PCNSL cases and 2-6% of all SCNSL cases [4-7]. SCNSL at the time of diagnosis is usually rare, and prognosis is worse compared to patients without CNS involvement [4, 9, 12]. In a study of 1,040 patients with PTCL, only two patients with PTCL-NOS (0.19%) presented with PCNSL, and 27 patients had SCNSL; 11 patients (1.06%) at the time of diagnosis and 16 patients (1.54%) at the time of relapse [4].
Based on retrospective studies, the risk factors for CNS involvement are identified as the presence of more than one extra nodal site, stage 3-4 disease, bone marrow involvement, and paranasal sinus disease. Symptoms of CNS disease include persistent headaches, focal neurological deficits (weakness or sensory impairments), new-onset seizures, and cognitive or behavioral changes. The brain is the most common site of involvement (> 90% of CNS cases), followed by the spinal cord, meninges, and peripheral neural plexus [6, 7, 21]. MRI is a sensitive imaging modality that usually shows a solitary mass or multiple lesions with enhancement, indicating disruption of the blood-brain barrier (BBB) (Table 2) [4, 6-8, 18-20].
Diagnosis of PTCL-NOS requires a combination of basic laboratory studies (LDH, complete blood count (CBC) and complete metabolic panel (CMP)), histopathological examination (lymph node biopsy showing diffuse infiltrates of atypical T cells with variable morphology), immunohistochemistry (to confirm T-cell origin and differentiate other lymphomas), flow cytometry and molecular genetic testing (to identify specific markers and genetic mutations), imaging studies (PET-CT, CT/MRI) and bone marrow biopsy (to identify sites of nodal and extra nodal disease) [3, 10, 13].
Standard frontline therapies for PTCL include CHOP (cyclophosphamide, doxorubicin, vincristine, prednisone), CHOEP (CHOP + etoposide), often used in younger patients with good performance status, and brentuximab vedotin + CHP (cyclophosphamide, doxorubicin, prednisone) [4, 8, 10, 22, 23]. Brentuximab vedotin has poor penetration of the BBB and is presumed to have poor efficacy in patients with CNS disease [8, 23]. High-dose methotrexate (HD-MTX, 3 to 8 g/m2) is standard in treating both primary and SCNSLs in PTCL due to its ability to penetrate the BBB. Monotherapy with HD-MTX demonstrated overall response rates of 35-74% in PCNSL. Combination therapies, HD-MTX with cytarabine and HD-MTX with rituximab, have shown improved complete response rates and median overall survival in both primary and SCNSL cases of PTCL. In patients with PTCL/PTCL-NOS at high risk for CNS involvement, prophylactic HD-MTX (3 g/m2) is administered during the initial treatment course to prevent CNS relapse [4, 8]. To mitigate toxicity, leucovorin is administered 24 h after HD-MTX infusion and is continued until methotrexate levels are deemed safe at < 0.1 µmol/L.
Consolidation with high-dose chemotherapy and autologous stem cell transplantation (ASCT) is recommended for eligible patients who achieve remission after initial chemotherapy [24]. Treatment options for relapsed or refractory (R/R) disease include pralatrexate (antifolate agent approved by the Food and Drug Administration (FDA)) [25], romidepsin (histone deacetylase inhibitor) [22], belinostat (histone deacetylase inhibitor) [26], as well as clinical trials investigating novel agents such as immune checkpoint inhibitors and targeted therapies like tyrosine kinase inhibitors (duvelisib) and JAK/STAT inhibitors [27]. In summary, the choice of treatment regimen should be individualized based on patient characteristics, lymphoma subtype, and treatment goals. Consultation with a multidisciplinary team is essential to optimize therapy and manage potential adverse effects.
In this report, we present a case of PTCL-NOS with CNS (brain parenchyma and meninges) and multi-nodal involvement at the time of diagnosis, which is an extremely rare presentation. PTCL-NOS is often aggressive and associated with a poor prognosis, and the IPI score is used to estimate the prognosis as noted above [10-12]. Another individual factor that influences survival is the response to initial therapy. Five-year overall survival and failure-free survival for all types of PTCL-NOS are 30-35% and 20%, respectively [10-13]. The prognosis for PTCL-NOS patients with CNS involvement is even worse, and the median survival time after CNS diagnosis is approximately 1.1 months [8, 12, 13].
Conclusions
PTCL-NOS is an aggressive disease with a varied clinical presentation. CNS involvement in PTCL-NOS is rare and can be primary or secondary. SCNSL at the time of diagnosis is uncommon, with the brain being the most frequent site of involvement, followed by the spinal cord and meninges. Strong predictors of CNS involvement and overall disease prognosis include an IPI score of ≥ 3, involvement of more than one extranodal site, and paranasal sinus disease.
In this case report, we present an extremely rare case of PTCL-NOS with multi-nodal involvement and SCNSL affecting the meninges at the time of diagnosis. CNS disease in PTCL/PTCL-NOS typically indicates a poor prognosis, with a median survival time of approximately 1.1 months following CNS diagnosis. Given its aggressive nature, early detection and prompt management are crucial for PTCL-NOS, especially in patients with CNS disease. Regimens that include high-dose methotrexate remain the standard treatment for PTCL/PTCL-NOS with CNS involvement. Further research and advancements are essential for understanding the disease’s behavior, pathogenesis, and developing more effective treatment strategies and targeted therapies.
Learning points
PTCL-NOS is an aggressive disease with rare CNS involvement, which could be primary or secondary. The brain is the most frequent site of CNS lymphoma in PTCL-NOS, followed by the spinal cord, meninges, and peripheral neural plexuses. CNS disease in PTCL-NOS typically indicates a poor prognosis, and early detection and prompt management are crucial for improved survival. Regimens that include high-dose methotrexate remain the standard treatment for PTCL-NOS with CNS involvement.
Acknowledgments
None to declare.
Financial Disclosure
The authors declare that they do not have a financial relationship with any commercial entity that is interested in the subject of this manuscript.
Conflict of Interest
The authors declare that they do not have any conflict of interest.
Informed Consent
This manuscript does not use patient identifiers or pictures of the patient. We obtained written informed consent from the patient’s next of kin (wife).
Author Contributions
All the authors participated actively in various sections of this manuscript prior to submission. Dr. Mrudula Thiriveedi (Primary author) started the case report, designed the article, actively wrote and edited multiple sections of the manuscript, including abstract, case report, discussion, and conclusions, conducted a literature review pertinent to the above sections, and gathered the images and pathology slides of the article. Dr. Muralidhar Idamakanti (Primary author) designed the article, actively wrote and edited multiple sections of the manuscript, including abstract, case report, discussion, and conclusions, conducted a literature review pertinent to the above sections, and performed language and grammatical editing. Drs. Siddharth Patel, Rafik ElBeblawy, Sujatha Baddam, and Bala Nimmana (Co-authors) participated in manuscript editing and literature review. Dr. Virginia Dailey contributed to the pathogenesis section, participated in the literature review, and obtained pathology slides. Dr. Rishi Patel (Co-author, corresponding oncologist) identified the rarity of the case, and participated in the article design, manuscript editing, and literature review.
Data Availability
The authors declare that data supporting the findings of this case report are available within the article.
Abbreviations
PTCL-NOS: peripheral T-cell lymphoma, not otherwise specified; NHL: non-Hodgkin lymphoma; CNS: central nervous system; PCNSL: primary CNS lymphoma; SCNSL: secondary CNS lymphoma; CT: computed tomography; MRI: magnetic resonance imaging; PET: positron emission tomography; FDG: fluorodeoxyglucose; ALK: anaplastic lymphoma kinase; HTLV-1: human T-lymphotropic virus-1; TET2: Tet methylcytosine dioxygenase 2; DNMT3A: DNA methyltransferase 3 alpha; IDH2: isocitrate dehydrogenase-2; JAK/STAT: Janus kinase-signal transducer and activator of transcription pathway; TBX: T-box; LDH: lactate dehydrogenase; CBC: complete blood count; CMP: complete metabolic panel; BBB: blood-brain barrier; HD-MTX: high-dose methotrexate; ASCT: autologous stem cell transplantation; R/R: relapsed or refractory; FDA: Food and Drug Administration
| References | ▴Top |
- Alaggio R, Amador C, Anagnostopoulos I, Attygalle AD, Araujo IBO,
Berti E, Bhagat G, et al. The 5th edition of the World Health Organization Classification of
haematolymphoid tumours: lymphoid neoplasms. Leukemia. 2022;36(7):1720-1748.
doi pubmed - Campo E, Jaffe ES, Cook JR, Quintanilla-Martinez L, Swerdlow SH,
Anderson KC, Brousset P, et al. The international consensus classification of mature lymphoid
neoplasms: a report from the clinical advisory committee. Blood. 2022;140(11):1229-1253.
doi pubmed - Vose J, Armitage J, Weisenburger D, International TCLP. International
peripheral T-cell and natural killer/T-cell lymphoma study: pathology findings and clinical
outcomes. J Clin Oncol. 2008;26(25):4124-4130.
doi pubmed - Mocikova H, Pytlik R, Benesova K, Janikova A, Duras J, Sykorova A,
Steinerova K, et al. Peripheral T-cell lymphomas involving the central nervous system: a report
from the czech lymphoma study group registry. Front Oncol. 2022;12:874462.
doi pubmed - Gurion R, Mehta N, Migliacci JC, Zelenetz A, Moskowitz A, Lunning M,
Moskowitz C, et al. Central nervous system involvement in T-cell lymphoma: A single center
experience. Acta Oncol. 2016;55(5):561-566.
doi pubmed - Chihara D, Oki Y. Central Nervous System Involvement in Peripheral T
Cell Lymphoma. Curr Hematol Malig Rep. 2018;13(1):1-6.
doi pubmed - Gandhi S, Bennani NN, Fortin S, Habermann TM, Johnston P, et al.
Central nervous system involvement in peripheral T-cell lymphoma. Blood.
2019;134(Supplement_1):5293.
doi - Lue JK, Ma H, Marchi E, Spivack JH, O'Connor OA. Peripheral T-cell
lymphoma involvement of the central nervous system: impact of novel therapeutics on clinical
outcomes. Blood. 2021;138 (Supplement 1):2469.
doi - Fuseya H, Nakao T, Hashimura M, Horiuchi M, Hayashi Y, Hagihara K,
Kanashima H, et al. [Peripheral T-cell lymphoma, not otherwise specified accompanied by central
nervous system involvement with features of lymphomatosis cerebri]. Rinsho Ketsueki.
2017;58(7):760-765.
doi pubmed - Weisenburger DD, Savage KJ, Harris NL, Gascoyne RD, Jaffe ES,
MacLennan KA, Rudiger T, et al. Peripheral T-cell lymphoma, not otherwise specified: a report of
340 cases from the International Peripheral T-cell Lymphoma Project. Blood.
2011;117(12):3402-3408.
doi pubmed - International Non-Hodgkin's Lymphoma Prognostic Factors P. A
predictive model for aggressive non-Hodgkin's lymphoma. N Engl J Med.
1993;329(14):987-994.
doi pubmed - Ma H, Marchi E, O'Connor OA, Lue JK. Mature T-cell and NK-cell
lymphoma involvement of the central nervous system: a single center experience. Leuk Lymphoma.
2023;64(12):1964-1970.
doi pubmed - Liu S, Liu W, Li H, Yang L, Song Y, Zhang X, Cheng Y, et al.
Epidemiological characteristics of peripheral T-cell lymphoma: a population-based study. Front
Oncol. 2022;12:863269.
doi pubmed - Yim J, Koh J, Kim S, Song SG, Bae JM, Yun H, Sung JY, et al.
Clinicopathologic and genetic features of primary T-cell lymphomas of the central nervous
system: an analysis of 11 cases using targeted gene sequencing. Am J Surg Pathol.
2022;46(4):486-497.
doi pubmed - Satou A, Takahara T, Tsuzuki T. Pathological and molecular features
of nodal peripheral T-cell lymphomas. Diagnostics (Basel). 2022;12(8):2001.
doi pubmed - Palomero T, Couronne L, Khiabanian H, Kim MY, Ambesi-Impiombato A,
Perez-Garcia A, Carpenter Z, et al. Recurrent mutations in epigenetic regulators, RHOA and FYN
kinase in peripheral T cell lymphomas. Nat Genet. 2014;46(2):166-170.
doi pubmed - Dogan A, Morice WG. Bone marrow histopathology in peripheral T-cell
lymphomas. Br J Haematol. 2004;127(2):140-154.
doi pubmed - Byun SH, Kim DM, Lee IH, Song CJ, Kim KH, Choi SY. Primary central
nervous system involvement in peripheral T-cell lymphoma: a case report. Taehan Yongsang
Uihakhoe Chi. 2021;82(1):255-260.
doi pubmed - Bird CE, Traylor JI, Thomas J, Caruso JP, Kafka B, Rosado F,
Blackburn KM, et al. Primary peripheral T-cell central nervous system lymphoma. Surg Neurol Int.
2021;12:465.
doi pubmed - Stark AM, Tiemann M, Dorner L, Melnikowa E, Mehdorn HM, Blomer U.
Primary peripheral T-cell lymphoma of the central nervous system. Zentralbl Neurochir.
2004;65(4):191-194.
doi pubmed - Kim EY, Kim SS. Magnetic resonance findings of primary central
nervous system T-cell lymphoma in immunocompetent patients. Acta Radiol.
2005;46(2):187-192.
doi pubmed - Coiffier B, Pro B, Prince HM, Foss F, Sokol L, Greenwood M, Caballero
D, et al. Romidepsin for the treatment of relapsed/refractory peripheral T-cell lymphoma:
pivotal study update demonstrates durable responses. J Hematol Oncol. 2014;7:11.
doi pubmed - Horwitz S, O'Connor OA, Pro B, Illidge T, Fanale M, Advani R,
Bartlett NL, et al. Brentuximab vedotin with chemotherapy for CD30-positive peripheral T-cell
lymphoma (ECHELON-2): a global, double-blind, randomised, phase 3 trial. Lancet.
2019;393(10168):229-240.
doi pubmed - d'Amore F, Relander T, Lauritzsen GF, Jantunen E, Hagberg H, Anderson
H, Holte H, et al. Up-front autologous stem-cell transplantation in peripheral T-cell lymphoma:
NLG-T-01. J Clin Oncol. 2012;30(25):3093-3099.
doi pubmed - O'Connor OA, Pro B, Pinter-Brown L, Bartlett N, Popplewell L,
Coiffier B, Lechowicz MJ, et al. Pralatrexate in patients with relapsed or refractory peripheral
T-cell lymphoma: results from the pivotal PROPEL study. J Clin Oncol.
2011;29(9):1182-1189.
doi pubmed - O'Connor OA, Horwitz S, Masszi T, Van Hoof A, Brown P, Doorduijn J,
Hess G, et al. Belinostat in patients with relapsed or refractory peripheral T-cell lymphoma:
results of the pivotal phase II BELIEF (CLN-19) study. J Clin Oncol.
2015;33(23):2492-2499.
doi pubmed - Zinzani PL, Zain J, Mead M, Casulo C, Jacobsen ED, Gritti G, et al.
P1172: Duvelisib in patients with relapsed/refractory peripheral t-cell lymphoma from the phase
2 primo trial: updated expansion phase analysis. Hemasphere. 2022;6(Suppl):1058-1059.
doi
This
article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0
International License, which permits unrestricted non-commercial use, distribution, and
reproduction in any medium, provided the original work is properly cited.
Journal
of Medical Cases is published by Elmer Press Inc.