| Journal of Medical Cases, ISSN 1923-4155 print, 1923-4163 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Med Cases and Elmer Press Inc |
| Journal website https://jmc.elmerpub.com |
Case Report
Volume 16, Number 5, May 2025, pages 187-193

A Case of High-Risk Myelodysplastic Syndrome With Cryoglobulinemia, Hemophagocytic Lymphohistiocytosis, and Progression to Multiple Organ Failure
Carlene A. Kranjaca, c ![]() , Linzi M. Hobbsb, Kavanya Feustelb, Karen-Sue Carlsonb
, Linzi M. Hobbsb, Kavanya Feustelb, Karen-Sue Carlsonb
aMedical College of Wisconsin, Milwaukee, WI, USA
bDivision of Hematology and Oncology, Department of Medicine, Medical College of Wisconsin, Milwaukee, WI, USA
cCorresponding Author: Carlene A. Kranjac, Medical College of Wisconsin, Milwaukee, WI 53226, USA
Manuscript submitted April 27, 2025, accepted May 13, 2025, published online May 28, 2025
Short title: High-Risk MDS With HLH and Cryoglobulinemia
doi: https://doi.org/10.14740/jmc5134
| Abstract | ▴Top |
Myelodysplastic syndromes (MDSs) are a group of hematological malignancies characterized by ineffective hematopoiesis. It is associated with genetic mutations, including p53 pathway genes, and can lead to complications, such as cytopenia and transformation to acute myeloid leukemia (AML). Hemophagocytic lymphohistiocytosis (HLH) is a rare, life-threatening condition that arises from immune dysregulation and often presents secondary to malignancies. Additionally, cryoglobulinemia, characterized by the precipitation of serum proteins at cooler temperatures, has been associated with infection, autoimmune disorders, and malignancies. A 59-year-old female recently diagnosed with high-risk MDS and a biallelic TP53 mutation presented to an outside hospital with persistent fevers. Initial evaluation revealed a Klebsiella pneumoniae urinary tract infection. Her condition rapidly deteriorated, and she developed acute kidney injury and respiratory failure, necessitating intensive care. She then developed HLH, indicated by elevated ferritin and CD25 levels despite a negative bone marrow biopsy for hemophagocytosis, which was then followed by cryoglobulinemia. The patient received corticosteroids for her HLH, plasmapheresis for her cryoglobulinemia, and a decitabine regimen for her MDS with gradual recovery of her organ function for a short time. She ultimately transformed to AML, requiring further intensive care before she passed away. The presence of a recently diagnosed high-risk MDS, HLH, cryoglobulinemia, and multi-organ failure emphasizes the complexity of this case. Despite meeting several diagnostic criteria for HLH, the patient’s bone marrow biopsy was negative for histiocytosis, emphasizing diagnostic challenges. The presence of cryoglobulinemia potentially linked to immune dysregulation further emphasizes the complexity of this case. While treatment with corticosteroids, plasmapheresis, and immunosuppressants provided stability, they did not cure her condition. Existing literature describes associations between high-risk MDS and HLH as well as MDS and cryoglobulinemia, but none addresses associations between all three processes. This case highlights an unusual occurrence of MDS, HLH, and cryoglobulinemia, emphasizing the need for awareness of the complex interactions between these conditions. Given the high-risk nature of her MDS and her unique clinical manifestations, further investigation into the underlying mechanisms driving these processes is necessary to enhance recognition and therapeutic approaches for affected patients.
Keywords: Myelodysplastic syndrome; Hemophagocytic lymphohistiocytosis; Cryoglobulinemia; Multiple organ failure
| Introduction | ▴Top |
Myelodysplastic syndromes (MDSs) are a group of hematological neoplasms in which the bone marrow undergoes ineffective or dysplastic hematopoiesis of erythrocytes, myeloid lineage leukocytes, and/or platelets [1]. According to the International Consensus Classification and World Health Organization (WHO) 2022 guidelines, MDS has been classified as 0-4% peripheral blasts or 5-19% bone marrow blasts. Additionally, a 20% blast count in the peripheral blood is no longer required for diagnosing AML with certain genetic abnormalities, and a new MDS/acute myeloid leukemia (AML) diagnosis was defined for blast counts ranging from 10% to 19% [2]. Exposure to alkylating chemotherapeutics, radiation, and environmental carcinogens, such as benzene, increases the risk of developing MDS. There is also a rare familial variant of MDS [1].
Genetic and epigenetic mutations that predispose to MDS include, from most to least common, splicing factors, deoxyribonucleic acid (DNA) methylation genes, histone modification genes, cohesin component genes, transcription factors, signal transduction genes, and p53 pathway genes [3].
Individuals with MDS may be asymptomatic for years before diagnosis. If symptoms present, they usually include anemia, fatigue, shortness of breath, chest pain, or dizziness [1]. Major complications of MDS are cytopenia or transformation to AML [1].
Hemophagocytic lymphohistiocytosis (HLH) is a rare and life-threatening condition that can arise, primarily from familial mutations, or secondary to infection, malignancy, rheumatological disease, and other underlying causes. It presents as an immune dysregulation due to increased activation of cytotoxic T lymphocytes, natural killer cells, and macrophages, leading to multiple end-organ failure [4]. In malignancy-associated cases, lymphoma is most commonly associated with HLH, with only a few cases reported of HLH secondary to MDS [4].
Cryoglobulinemia can result from infection, such as hepatitis C, autoimmune diseases, such as systemic lupus erythematosus, and various malignancies [5]. Cryoglobulinemia are proteins, specifically cryoglobulins, that aggregate from an individual’s plasma at cooler temperatures, less than 37 °C [5]. There are three types of cryoglobulinemia: type I is a hemostasis disorder with signs of medium-vessel vasculitis whereas type II and type III are mixed cryoglobulinemia disorders characterized by small-vessel vasculitis and complement-mediated immune-complex deposition [6]. Type I cryoglobulinemia is often associated with hematological malignances, whereas type II and type III cryoglobulinemia are often associated with hepatitis C infection. Common findings in type I cryoglobulinemia typically include increased levels of IgG, undetectable rheumatoid factor activity, and complement levels within normal limits. Skin biopsies, when done, will often demonstrate noninflammatory thrombotic lesions with infarction or hemorrhage. Type II cryoglobulinemia will typically have increased levels of IgM, increased rheumatoid factor levels, and decreased complement levels, whereas these findings can be inconsistent in type III cryoglobulinemia. However, both types II and III will typically present with a leukocytoclastic vasculitis and thrombi on skin biopsy [6].
This paper discusses a patient diagnosed with high-risk MDS with a biallelic TP53 mutation who rapidly developed HLH, cryoglobulinemia, and multisystem organ failure.
| Case Report | ▴Top |
In September 2024, a 59-year-old female with recently diagnosed high-risk MDS and a past medical history of overactive bladder, peripheral neuropathy, anxiety, and past tobacco use presented to an outside hospital with 3 days of persistent fevers to 103 °F (39.44 °C) at home and 3 weeks of dry cough, headache, and sore throat. She denied chest pain, shortness of breath, dysuria, edema, and abdominal pain.
Prior to this admission, she was diagnosed with MDS (WHO5/ICC-2022) after lab work demonstrated macrocytic anemia with macrocytosis, atypical granulocytes, hypogranulation, and hyposegmentation on a peripheral blood smear. Bone marrow biopsy demonstrated hypercellular marrow (60%) with multilineage dysplasia with less than 10% bone marrow blasts and no circulating blasts, and cytogenetics confirmed MDS with a biallelic TP53 mutation at the 17p13 chromosome; these features are commonly associated with aggressive disease and a poor prognosis [3]. She was scheduled to start chemotherapy in September 2024 but presented to the emergency department before this date.
On admission to the outside hospital, her D-dimer was elevated to 124,000, but her chest X-ray showed no acute cardiopulmonary disease. Clinical and diagnostic workup indicated no signs of pulmonary embolism or airspace disease on computed tomography (CT) pulmonary embolism and bilateral lower extremity Dopplers. The infectious respiratory panel was negative. A urinalysis was positive for bacteria and urine culture grew Klebsiella pneumoniae. She was started on a 14-day regiment of ceftriaxone for possible sepsis secondary to bacterial urinary tract infection.
Two days later, she developed another fever of 101.2 °F (38.44 °C) alongside new shortness of breath, wheezing, and hypoxia. Chest X-ray at this time demonstrated diffuse reticular interstitial opacities that could not exclude congestive processes or infectious etiologies. Bilateral upper extremity venous Dopplers ruled out deep venous thrombosis. A CT scan of her abdomen and pelvis, along with elevated lipase levels confirmed pancreatitis. She was treated with intravenous (IV) fluids and bowel rest, but her hospitalization was further complicated by oliguric acute kidney injury with pulmonary edema and hypoxemic respiratory failure, leading to rapid progression with bilevel positive airway pressure (BiPAP). Her creatinine had risen to 4.75, from a baseline of 0.8, requiring hemodialysis.
Additionally, she presented with new-onset atrial fibrillation with rapid ventricular response, controlled with medication management. An echocardiogram demonstrated ejection fraction (EF) 55-60% and normal ventricular function. Her CHA2DS2-VASc score was 2 - 3. With no clear etiology for her fever, along with the rapid kidney and respiratory failure, the outside hospital requested a transfer to a tertiary care center for a higher level of care.
On arrival at the tertiary care center, she was admitted to the intensive care unit (ICU), thought to be experiencing systemic inflammatory response syndrome (SIRS) secondary to her idiopathic pancreatitis. A complete infectious workup was initiated and negative for Aspergillus species, Streptococcus species, coronavirus disease 2019 (COVID-19), hepatitis, and tick-borne illnesses, such as Lyme disease. Her C-reactive protein (CRP) was elevated to 416 (< 10.0 mg/L) and ferritin was 2,500 (18.0 - 340.0 ng/mL). She was saturating at 95% oxygen on a 10-L nasal cannula (NC), creatinine was 3.54 (0.50 - 1.10 mg/dL), and potassium was 4.1 (3.4 - 5.1 mmol/L). Her anion gap was 24 (7 - 15 mmol/L), thought to be due to renal failure or diabetic ketoacidosis. However, her anion gap did not improve with an insulin drip and the patient continued hemodialysis. She was suffering from stage 3 acute kidney injury, likely secondary to sepsis, pancreatitis, hemodynamic instability related to her atrial fibrillation, and IV contrast use.
Additionally, her bilateral toes were cooler than normal with diminished pulses, raising concern for cryoglobulinemia. Initial workup for cryoglobulinemia was negative; she had elevated kappa and lambda free light chains, but the ratio was within normal limits at 1.35 (0.40 - 2.58). Epstein-Barr virus (EBV) testing was negative and bilateral lower extremity arterial Dopplers showed no evidence of occlusion or stenosis.
Due to this constellation of symptoms and multi-organ failure, HLH was on the differential. Her initial H-score was 155, indicating a 25-40% chance of HLH; her fibrinogen was 520 (193 - 507 mg/dL), ferritin was 5,034 (18.0 - 340.0 ng/mL), aspartate aminotransferase (AST) was 85 (< 35 U/L), and CD25 serum test was 4,271.2 (175.3 - 858.2 pg/mL). The repeat H-score was 142, indicating a 16-25% risk. Additionally, there was no hemophagocytosis on her bone marrow biopsy in August 2024.
Two days later, she was weaned down to 2 L oxygen NC and eventually room air and transferred to the floor with worsening cyanosis of her bilateral toes. Heparin was started for anticoagulation.
The following day, her platelets dropped to 25 × 103/µL from 32 × 103/µL (165 - 366 × 103/µL) with no hemolysis, confirmed by a lack of spherocytes, schistocytes, bite cells, reticulocytosis, or polychromasia, on blood smear, raising concern for heparin-induced thrombocytopenia (HIT). HIT antibody testing was negative. Notably, at this time, she developed purpura on her shins. Biopsy of these purpuric lesions demonstrated microthrombi and microvascular vasculitis.
On day 4, she began to hyperventilate and experience altered mental status. She was tachycardic with fevers of 102 °F (38.89 °C). Venous blood gases came back with a pH of 7.40 (7.32 - 7.42), pCO2 of 32 (42 - 55 mm Hg), and HCO3 of 40 (23 - 33 mmol/L). A rapid response was called, and she was transferred back to the ICU. She was intubated the next day, started on pressors, and sedated. IV methylprednisolone was begun to address the possible inflammation that she has been having. Plasma exchange was also initiated to treat her presumed cryoglobulinemia.
On day 7, she was extubated and transferred back to the floor the following day. Rheumatological etiologies were considered, and lab work was negative except for a positive lupus anticoagulant (Table 1). The patient was then started on a 5-day regimen of decitabine for her MDS and a steroid taper for HLH. At the end of her decitabine, she was started on once-weekly rituximab infusions for 4 weeks to help with the cryoglobulinemia while vascular surgery was planned for possible amputation.
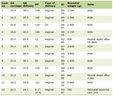 Click to view | Table 1. Laboratory Values |
On her day of discharge, she was still receiving hemodialysis three times per week, but her kidney function was improving as seen by an increase in urine output and a gradual return of her creatinine to her baseline. The cryoglobulinemia in her toes was demarcating, and she slowly started to gain sensation and pain back in one foot. She was discharged to an inpatient rehabilitation facility with continued hemodialysis and weekly rituximab (for a total of four doses).
The patient, unfortunately, presented back to the hospital 2 days later with diverticulitis and C. difficile infection. The hospitalization was complicated by atrial fibrillation with rapid ventricular response requiring a transfer to the ICU for an amiodarone infusion. She was transferred back to the floor and then shortly moved back to the ICU for continuous renal replacement therapy, as her renal function worsened, and also for tachypnea requiring intubation. The patient started cycle two of decitabine while in the ICU but continued to worsen. Her blast percentage increased to 43% (0%); it was likely that her MDS had progressed to AML. Given the rapid decline of the patient and her poor prognosis, the patient passed away.
| Discussion | ▴Top |
About 20% of MDS patients harbor the TP53 mutation, which is associated with worsened health outcomes as compared to other MDS mutations, such as splicing factors, DNA methylation genes, histone modification genes, cohesin component genes, transcription factors, and signal transduction genes [3]. Notably, existing literature mentions no associations between this high-risk MDS mutation and the development of HLH. This case presents a unique instance of a patient with recently diagnosed MDS who rapidly progressed to multiple end-organ failure and developed HLH, highlighting the complex interactions between these conditions.
The HLH-2004 criteria require fulfillment of five out of eight criteria for an HLH diagnosis. These criteria were created by the Histiocyte Society in a pediatric population with primary HLH and include: fever > 38.5 °C/101.3 °F, splenomegaly, cytopenia affecting two or more lineages in peripheral blood (hemoglobin < 9 g/dL, platelets < 100 × 109/L, and neutrophils < 1.0 × 109/L), hypertriglyceridemia (> 265 mg/dL) or fibrinogen < 1.5 g/L, hemophagocytosis in the bone marrow, spleen, liver, lymph nodes, or other tissue, low or absent natural killer cell activity, serum ferritin concentration > 500 µg/L, and soluble CD25 > 2,400 U/mL [7].
Due to the HLH-2004’s unclear diagnostic value in secondary HLH, the H-score was developed in 2014 for adults with secondary HLH. The criteria for this score include known underlying immunosuppression, fever, organomegaly, cytopenia, ferritinemia, triglyceridemia, fibrinogenemia, elevated AST, and hemophagocytosis in bone marrow [8].
This patient met many of these criteria; however, the exact etiology of this patient’s HLH remains unclear. MDS drives chronic inflammation through a combination of innate immune dysregulation, the bone marrow microenvironment, and genetic mutations. In MDS, the innate immune system is chronically activated due to increased levels of inflammatory cytokines, such as tumor necrosis factor-alpha (TNF-α) and interleukin-1 (IL-1), contributing to the ineffective hematopoiesis within the bone marrow that defines the disease [9]. One of these mechanisms involves the S100A9-mediated NOD-like receptor protein 3 (NLRP3) inflammasome, which activates pyroptosis, an inflammatory form of cell death, and ultimately creates a positive feedback effect that further amplifies inflammasomes and pro-inflammatory cytokines, such as IL-18 and IL-1B, further activating the innate immune system [9, 10]. Additionally, genetic mutations, such as RNA splicing and DNA methylation mutations, have the ability to enhance the expression of pro-inflammatory mediators, facilitating an inflammatory microenvironment [11]. A study from 2021 investigated the impact of the SF3B1 gene in patients with MDS. They found that inflammatory cytokines were significantly elevated and led to a pro-inflammatory environment that ultimately led to worsened patient outcomes [12]. This increased inflammation could lead to the development of HLH, as the New England Journal of Medicine classifies this disease as a syndrome of hyperinflammation [13]. In this patient’s case, elevated CD25 and ferritin levels, along with persistent fevers and evidence of end-organ damage point to HLH, possibly secondary to her MDS; however, it is noteworthy that her bone marrow biopsy was negative for histiocytosis. While we recognize the limitations to this interpretation, the exclusion of other etiologies on laboratory workup (Table 1), including infectious, rheumatological, and hematological, supports the plausibility of an MDS etiology.
Although HLH is known for a wide variety of symptoms, its presence in a recently diagnosed high-risk MDS is uncommon. While HLH symptoms commonly include fever, organomegaly, hypotension, elevated ferritin, and cytopenia, this patient experienced rapid progression to multiple organ failure and cryoglobulinemia [4]. These findings underscore the need for increased awareness of HLH in patients with MDS, particularly those with a systemic inflammatory response and rapid decline in organ function.
Moreover, the presence of cryoglobulinemia-like symptoms in the setting of trace cryoglobulins emphasizes the complexity of this presentation. The etiology remains unknown and the association of cryoglobulinemia with MDS and HLH is uncommon. In cases where it is hard to identify cryoglobulins, as in the case of this patient, the presence of cryoglobulinemia can also be made with hypergammaglobulinemia, low complement levels, and increased rheumatoid factor activity [6]. In this case, the patient had immunoglobulins, complement levels, and rheumatoid factor within normal limits, suggesting that these cryoglobulinemia-like symptoms could be a type I cryoglobulinemia, which is associated with hematological malignancy, but also may be a mimic of cryoglobulinemia [6]. Studies show that other medical conditions, such as systemic lupus erythematous, infection, hematological disorders, and other autoimmune disorders can present with symptoms that present as cryoglobulinemia despite trace cryoglobulins and other laboratory values within normal limits [6]. This patient had a positive lupus anticoagulant test that could indicate the presence of an antiphospholipid syndrome which could also be a driver of these cryoglobulinemia-like symptoms. However, studies have found that lupus anticoagulants can be falsely positive with the use of medications, such as heparin, and direct oral anticoagulants, like argatroban and rivaroxaban [14]. This patient was taking both heparin and enoxaparin at various times throughout her hospitalization. Additionally, this patient had negative anticardiolipin antibodies and anti-beta2-glycoprotein I antibodies and has not received a second lupus anticoagulant panel yet, keeping antiphospholipid syndrome on the differential [14]. Moreover, the microthrombi and microvascular vasculitis found on this patient’s skin biopsy, alongside the cyanosis of her bilateral toes after her rapid decline in health potentially suggests that immune dysregulation in MDS and HLH could contribute to the development of cryoglobulinemia-like symptoms. The unclear etiology of her symptoms identifies the need to better understand the pathophysiology and the role of inflammation between these disorders.
The treatment for patients with MDS, HLH, and cryoglobulinemia is not well understood and requires a multi-faceted approach. In this case, the patient was treated with corticosteroids, plasmapheresis, supportive care for her organ dysfunction, and immunosuppressants, such as rituximab. While this treatment regimen did not cure her MDS, HLH, or cryoglobulinemia, these medications provided stability and allowed her organs to gradually regain function for a short time, and also for her cryoglobulinemia to improve.
There is minimal existing literature demonstrating associations between MDS, HLH, and cryoglobulinemia. A retrospective study from 2023 investigated 15 cases of MDS associated with HLH at a single hospital. This study found that high-risk MDS patients often developed HLH during MDS progression or relapse, while low-risk individuals developed HLH during the time of MDS onset [15]. Additionally, in a case report and literature review from 2018, authors describe the difficulty in treating HLH due to MDS, emphasizing the importance of understanding the differences in pathophysiology that exist with MDS associated with HLH versus other malignancies [16]. Finally, a retrospective study from 2011 looked at data for 235 patients with MDS and found cryoglobulins in 54% of the cases. This study demonstrated that the detection of cryoglobulins in MDS patients was associated with worsened health outcomes [17].
Conclusion
This is a unique case that describes the simultaneous manifestations of MDS, HLH, and cryoglobulinemia. This case seeks to encourage further exploration into the underlying mechanisms between these disorders and the etiologies that fuel their development. In this case, MDS appears to be a driver for HLH; however, the role of either MDS or the presence of a positive lupus anticoagulant in the progression of cryoglobulinemia-like symptoms remains elusive. An increased understanding of these associations could significantly enhance the recognition and therapeutic management for improved patient outcomes.
Acknowledgments
We extend our gratitude to the patient and their family for allowing us to share this case to contribute to medical knowledge.
Financial Disclosure
None to declare.
Conflict of Interest
None to declare.
Informed Consent
Informed consent was obtained by the patient prior to her passing for publication of this report.
Author Contributions
Carlene A. Kranjac, BS, Linzi Hobbs, MD, Kavanya Feustel, MD, and Karen-Sue Carlson, MD, PhD: substantial contributions to the conception or design of the work; or the acquisition, analysis, or interpretation of data for the work; drafting the work or revising it critically for important intellectual content; final approval of the version to be published; agreement to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Data Availability
The authors declare that data supporting the findings of this study are available within the article.
Abbreviations
AML: acute myeloid leukemia; BiPAP: bilevel positive airway pressure; COVID-19: coronavirus disease 2019; CRP: C-reactive protein; CT: computed tomography; DNA: deoxyribonucleic acid; EBV: Epstein-Barr virus; EF: ejection fraction; HIT: heparin-induced thrombocytopenia; HLH: hemophagocytic lymphocytosis; ICU: intensive care unit; MDS: myelodysplastic syndrome; NC: nasal cannula; SIRS: systemic inflammatory response syndrome; WHO: World Health Organization
| References | ▴Top |
- Dotson JL, Lebowicz Y. Myelodysplastic syndrome. In: StatPearls. Treasure Island (FL) ineligible companies. 2025.
pubmed - M Hamed NA. Classification of myelodysplastic neoplasms in the 5th edition of the World Health Organization classification compared to 2022- International Consensus Classification. Cancer Therapy & Oncology International Journal. 2023;23(5):556135.
doi - Haferlach T. The Molecular pathology of myelodysplastic syndrome. Pathobiology. 2019;86(1):24-29.
doi pubmed - Ponnatt TS, Lilley CM, Mirza KM. Hemophagocytic lymphohistiocytosis. Arch Pathol Lab Med. 2022;146(4):507-519.
doi pubmed - Killeen RB, Awais M, Mikes BA. Cryoglobulinemia. In: StatPearls. Treasure Island (FL) ineligible companies. 2025.
pubmed - Cacoub P, Vieira M, Saadoun D. Cryoglobulinemia - one name for two diseases. N Engl J Med. 2024;391(15):1426-1439.
doi pubmed - Zhang K. Table 1. HLH-2004 diagnostic criteria. genereviews® - NCBI bookshelf. GeneReviews® [Internet]. June 6, 2024. Accessed October 11, 2024. https://www.ncbi.nlm.nih.gov/books/NBK1444/table/hlh.T.hlh2004_diagnostic_criteria/.
- Fatma A, Raida BS, Mourad C, Ikram D, Zouheir B, Henda E. Performances of the H-score and the HLH-2004 score in the positive diagnosis of secondary hemophagocytic lymphohistiocytosis. Curr Res Transl Med. 2024;72(2):103430.
doi pubmed - Sallman DA, List A. The central role of inflammatory signaling in the pathogenesis of myelodysplastic syndromes. Blood. 2019;133(10):1039-1048.
doi pubmed - Basiorka AA, McGraw KL, Eksioglu EA, Chen X, Johnson J, Zhang L, Zhang Q, et al. The NLRP3 inflammasome functions as a driver of the myelodysplastic syndrome phenotype. Blood. 2016;128(25):2960-2975.
doi pubmed - Matos A, Magalhaes SMM, Rauh MJ. Immune dysregulation and recurring mutations in myelodysplastic syndromes pathogenesis. Adv Exp Med Biol. 2021;1326:1-10.
doi pubmed - Pollyea DA, Kim HM, Stevens BM, Lee FF, Harris C, Hedin BR, Knapp JR, et al. MDS-associated SF3B1 mutations enhance proinflammatory gene expression in patient blast cells. J Leukoc Biol. 2021;110(1):197-205.
doi pubmed - Henter JI. Hemophagocytic Lymphohistiocytosis. N Engl J Med. 2025;392(6):584-598.
doi pubmed - Favaloro EJ, Pasalic L, Selby R. Testing for the lupus anticoagulant: the good, the bad, and the ugly. Res Pract Thromb Haemost. 2024;8(3):102385.
doi pubmed - Song Y, Zhou F, Li X, et al. Myelodysplastic syndrome associated hemophagocytic lymphohistiocytosis: A retrospective study of 15 cases in a single center. Blood. 2023;142(Supplement 1):1167.
doi - Sun Y, Blieden C, Merritt BY, Sosa R, Rivero G. Hemophagocytic lymphohistiocytosis and myelodysplastic syndrome: a case report and review of the literature. J Med Case Rep. 2021;15(1):98.
doi pubmed - de Hollanda A, Beucher A, Henrion D, Ghali A, Lavigne C, Levesque H, Hamidou M, et al. Systemic and immune manifestations in myelodysplasia: a multicenter retrospective study. Arthritis Care Res (Hoboken). 2011;63(8):1188-1194.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Medical Cases is published by Elmer Press Inc.