| Journal of Medical Cases, ISSN 1923-4155 print, 1923-4163 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Med Cases and Elmer Press Inc |
| Journal website https://jmc.elmerpub.com |
Case Report
Volume 000, Number 000, February 2025, pages 000-000

Hemophagocytic Lymphohistiocytosis After Treatment With Checkpoint Inhibitor Therapy
Cameron Peresa, Christopher Willnera, b
aDivision of Hematology and Oncology, Henry Ford Cancer Institute, Detroit, MI 48202, USA
bCorresponding Author: Christopher Willner, Division of Hematology and Oncology, Henry Ford Cancer Institute, Detroit, MI 48202, USA
Manuscript submitted August 21, 2024, accepted October 21, 2024, published online February 18, 2025
Short title: HLH Disease With Immune Checkpoint Inhibitor
doi: https://doi.org/10.14740/jmc4318
| Abstract | ▴Top |
Hemophagocytic lymphohistiocytosis (HLH) is a rare hematological syndrome presenting with massive, dysregulated cytokine release that can result in multiple organ failure and is associated with a high risk of mortality. Based on the recent North American consortium recommendations, it has been suggested to categorize HLH into two entities, HLH syndrome and HLH disease. HLH disease encompasses multiple subgroups, including familial HLH (F-HLH), HLH-associated immune compromise (IC-HLH) and HLH observed after immune activating therapies. The diagnosis can be quite challenging, and the pathophysiology leading to HLH disease has yet to be fully elucidated. Much less is known about HLH that occurs due to treatment with immunotherapy such as immune checkpoint inhibitors (ICIs). Herein, the authors report a case of a 71-year-old man who was treated with a combination of nivolumab and ipilimumab for bladder cancer. He later presented with mental status changes and pancytopenia, ultimately meeting the diagnostic criteria for HLH syndrome.
Keywords: HLH; Checkpoint inhibitor; Cancer
| Introduction | ▴Top |
Hemophagocytic lymphohistiocytosis (HLH) is a rare and severe hyper-inflammatory syndrome. Most recently, based on the recommendations from the North American Consortium for Histiocytosis (NACHO), HLH is classified into HLH syndrome or HLH disease [1]. HLH disease can be sub-grouped into familial HLH (F-HLH) with a genetic etiology, HLH associated with malignancy (M-HLH), HLH after immune activating therapies and HLH associated with an immune-compromised state (IC-HLH) [1].
The pathophysiology of F-HLH has been studied in animal models and is caused by the excessive activation of CD8+ T cells, with the upregulation of interferon gamma (IFN-γ) being a key mediator of disease [2]. Most of the mutations affecting perforin delivery, such as defects in PRF1, UNC13D and STX11, have been identified as causes of F-HLH [2].
HLH syndrome is defined by meeting the consensus of diagnostic criteria and those conditions that would benefit from HLH-directed therapies [1, 2]. The majority of HLH syndrome cases are caused by a genetic mutation affecting the function of the T lymphocytes and natural killer (NK) cells in F-HLH, with an incidence of 1 to 225 per 300,000 live births [1, 2]. M-HLH has been recognized for decades and can occur prior to the diagnosis or during the treatment of the malignancy [3]. When M-HLH presents in a newly diagnosed patient, it is usually driven by the cancer, with the recommended therapy being that for the specific neoplasm itself [3]. M-HLH used to have a very poor prognosis; however, with newer therapeutic options and early recognition, survival rates have improved [3]. The incidence of M-HLH is estimated to be about 1%. With the advent of immune checkpoint inhibitors (ICIs), bispecific monoclonal antibodies, and bispecific T-cell engagers (BiTE), as well as chimeric antigen receptor T-cell (CAR-T) therapies, the incidence of immune-related adverse events (irAEs) is being increasingly reported [4].
Diagnosing HLH is based on the criteria established by the Histiocyte Society in 1994, with a revision in 2004. Criteria include persistent fever, bilineage or trilineage cytopenias, splenomegaly, elevated ferritin, hypofibrinogenemia, increased soluble CD25 (sCD25) and hemophagocytosis (Table 1) [1-4]. Recently, HLH has been described as a potential irAE associated with ICI [5].
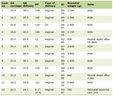 Click to view | Table 1. HLH-2004 Diagnostic Criteria |
Immunotherapy has shown significant promise in the treatment of various cancers. It has improved overall survival in several different types of both hematologic malignancies and solid tumors [6]. The following is a case report of HLH associated with ICI therapy.
| Case Report | ▴Top |
The patient was a 71-year-old man who was undergoing therapy for small cell carcinoma of the bladder, treated with ipilimumab and nivolumab. He presented with fever and encephalopathy of 2-day duration following his initial cycle. He had not sustained head trauma, loss of consciousness, sore throat, cough, abdominal pain, diarrhea, dysuria or sick contacts. Examination revealed a heart rate of 121 beats per minute, a temperature of 101.4 °F, tachypneic and only alert to person. Laboratory workup was significant for a white blood cell count of 2.6 × 103/µL (normal range: 3.8 - 10.6 × 103/µL), hemoglobin of 9.1 g/dL (normal range: 12.0 - 15.0 g/dL) and platelet count of 18 × 103/µL (normal range: 150 - 450 × 103/µL). Triglycerides were 280 mg/dL (normal range: 30 - 150 mg/dL), ferritin was 10,058 ng/mL (normal range: 11 - 307 ng/mL) and creatinine 1.25 mg/dL (normal range: < 1.16 mg/dL). He was treated with broad-spectrum antibiotics, and an infectious workup, including a hepatitis viral panel, fungal cultures, and additional viral studies, were completed. Computed tomography imaging of the chest, abdomen, and pelvis revealed splenomegaly and lymphadenopathy. Peripheral blood smear revealed moderate normochromic anemia, leukopenia with lymphopenia and left-shifted neutrophils. There was marked thrombocytopenia, though no schistocytes, dysplasia or increased blasts were observed. Based on this, other causes of fever were excluded, including hematological and rheumatological causes. Having met the criteria, including fever, splenomegaly, hypertriglyceridemia, starkly elevated ferritin and pancytopenia, HLH was considered. He was started on treatment with corticosteroids and HLH therapy, however, his symptoms worsened, leading to progressive multiple organ failure, and he expired.
| Discussion | ▴Top |
We describe a case of a 71-year-old man who was undergoing therapy for small cell carcinoma of the bladder and was treated with ipilimumab and nivolumab. He presented with fever, encephalopathy, pancytopenia, splenomegaly, elevated ferritin and hypertriglyceridemia. These symptoms meet the criteria for a diagnosis of HLH, most likely due to the ICI therapy that he received, which was unique to this case. He progressed rapidly over 3 to 4 days and developed multiple organ failure without additional HLH-directed therapy.
HLH is a clinical syndrome that has been recognized since the early 1950s and is characterized by the uncontrolled activation of immune cells, which leads to systemic hyperinflammation and multiple organ failure [7]. The activation of CD8+ lymphocytes and macrophages results in multiple organ damage in the central nervous system and bone marrow [7]. Even though the pathophysiology is unique and has been defined in animal models of F-HLH, the diversity of clinical presentations in patients with HLH remains ambiguous. With the recently revised NACHO recommendations, M-HLH is usually an HLH mimic, and the features of this syndrome are driven by the underlying malignancy [8]. There is a great diversity of HLH presentations, which tend to be heterogeneous in nature. A descriptive analysis of patients diagnosed with HLH, who subsequently were identified to have underlying neoplasms, reported that less than 50% of adults with M-HLH received HLH-directed treatment. This was due to the lack of the diagnosis in adult patients with malignancies, or those who are receiving treatment for their malignancies [8]. ICI promotes antitumor activity by blocking down-regulators of immunity, like anti-cytotoxic T-lymphocyte antigen 4 (anti-CTLA-4) and the programmed cell death protein 1 (PD-1) [9]. The mechanism of action of ICI is dependent on the ability to inhibit barriers of immune activation. However, they could potentially also cause immune-mediated inflammation of various organ systems [9]. A systematic review by Rajapakse et al reported that of 22 patients who developed HLH while receiving treatment with an ICI, it was found that fever was the most common symptom at presentation (90.9%), with the most common laboratory findings being cytopenias and elevated ferritin (90.9%) [10]. A bone marrow examination was obtained in 19 (86.36%) cases, which revealed features of hemophagocytosis. Although other potential causes of HLH, including infection and progressive neoplastic disease, were considered in all cases, there was no sufficient evidence to confirm the association between the other causes of HLH [10]. Thus, it was concluded that HLH was related to recipients of ICI therapy, suggesting that these cases may be underrecognized and supporting further investigation that might elucidate a clear pathophysiologic relationship. One such hypothesis is that patients with malignancy already have a compromised immune system, which could lead to a disrupted immune pathway, including macrophage hyperactivation and unchecked CD8+ cellular activation by ICI [11].
The pathogenesis of HLH is not well understood and can be based on experimental studies in animal models. There have been several proposed mechanisms, including the stimulation of toll-like receptors (TLRs) leading to excessive activation of the innate immune system [12]. Epstein-Barr virus, which is one of the most common infectious triggers, followed by cytomegalovirus, engages the TLR9 to cause stimulation of the myeloid cells [12, 13]. Interleukin-18 (IL-18) overproduction by activated epithelial and myeloid cells is also a possible mechanism leading to the amplification of IFN-γ, which is the key mediator of disease development [12, 13]. Irrespective of the pathogenesis of HLH, the excessive activation of CD8+ T cells leads to the excessive production of inflammatory cytokines [13]. A recent study by De Matteis et al revealed that highly activated CD38 high/HLA-DR+CD8+ T cells are expanded in patients with HLH, and that CD4 dim CD8+ cells are significantly increased in patients with HLH, regardless of the trigger or underlying condition [14].
Immediate treatment of HLH is warranted once a diagnosis is made. The treatment of M-HLH should start by targeting the trigger regarding the malignancy itself. It is generally accepted that treatment of the underlying neoplasm will attenuate malignancy-related HLH manifestations. However, continued close inpatient monitoring while assessing the need to start treatment for hyperinflammation must be taken into consideration. The most commonly used regimen, based on the modified HLH-94 protocol and including the combination of dexamethasone and etoposide, was given in a prospective trial initiated by the Histiocyte Society in 1994 [15-17]. Cyclosporine was subsequently added during the continuation phase of treatment. Resolution of HLH was observed in 53% of patients, improvement was observed in an additional 32%, and no improvement was observed in 4% of the patients [17]. Despite HLH treatment, the prognosis remains poor, with the overall survival being 61% at 5 years, based on the HLH-2004 study [17].
Immunosuppression targeting individual cytokines has also been studied. Anakinra, an interleukin-1 (IL-1) receptor antagonist, has been studied retrospectively in 44 patients with HLH with a 73% survival rate [4, 18, 19]. Tocilizumab, an anti-interleukin-6 receptor (IL-6R) antibody, has been used in patients undergoing CAR-T therapy for cytokine release syndrome, and, owing to the homology of the cytokine pattern between cytokine release syndrome (CRS) and HLH, it has been postulated that it might be beneficial in inducing remission in patients with HLH [4, 19]. Our patient was treated with dexamethasone, but due to the rapid progression of HLH and multiple organ failure, he expired before additional HLH therapy could be started.
Conclusions
Our case adds to the base of literature that suggests HLH may occur in relation to immunotherapy and is associated with significant morbidity and a high likelihood of mortality. The high mortality in this patient population could be due to multiple factors, including patients who are immunosuppressed, delays in recognizing the disorder, and overall response to treatment. Advancements in the treatment of HLH have dramatically improved the outcomes for patients, with long-term remissions and cures being possible in a proportion of patients. However, early mortality for this disease remains high, and the need for prompt recognition justifies the need to report and establish a potential relationship between HLH and ICI therapy. Further studies are needed in patients who experience HLH associated with ICI therapy to better define the best therapeutic options and improve the overall outcomes of this rare complication. Future directions could include identifying potential biomarkers that could help further assist in making the diagnosis sooner and potentially starting therapy sooner to reduce mortality.
Learning points
This case highlights the importance of patients being treated with ICI therapy for an underlying malignancy that can go on to develop HLH. It remains a rare but serious irAE associated with ICI therapy. Our case reinforces the importance of early recognition, diagnosis and treatment of ICI-induced HLH due to its potentially fatal course in patients with underlying malignancy.
Acknowledgments
We would like to thank Dr. Zaher Otrock for his review of this manuscript, and Analise Johnson, MLIS, for her review as well.
Financial Disclosure
None to declare.
Conflict of Interest
The authors declare that there is no conflict of interest regarding the subject matter of this paper.
Informed Consent
Institutional guidelines were followed, and informed consent was obtained.
Author Contributions
Christopher Willner contributed to the study conception, design and data collection of the article. Cameron Peres contributed to the data collection, analysis and interpretation of the results. All authors reviewed the draft manuscript, including results, and approved the final version of the manuscript.
Data Availability
The authors declare that data supporting the findings of this study are available within the article.
| References | ▴Top |
- Jordan MB, Allen CE, Greenberg J, Henry M, Hermiston ML, Kumar A, Hines M, et al. Challenges in the diagnosis of hemophagocytic lymphohistiocytosis: Recommendations from the North American Consortium for Histiocytosis (NACHO). Pediatr Blood Cancer. 2019;66(11):e27929.
doi pubmed - Stepp SE, Dufourcq-Lagelouse R, Le Deist F, Bhawan S, Certain S, Mathew PA, Henter JI, et al. Perforin gene defects in familial hemophagocytic lymphohistiocytosis. Science. 1999;286(5446):1957-1959.
doi pubmed - Lehmberg K, Sprekels B, Nichols KE, Woessmann W, Muller I, Suttorp M, Bernig T, et al. Malignancy-associated haemophagocytic lymphohistiocytosis in children and adolescents. Br J Haematol. 2015;170(4):539-549.
doi pubmed - La Rosee P, Horne A, Hines M, von Bahr Greenwood T, Machowicz R, Berliner N, Birndt S, et al. Recommendations for the management of hemophagocytic lymphohistiocytosis in adults. Blood. 2019;133(23):2465-2477.
doi pubmed - Bode SF, Ammann S, Al-Herz W, Bataneant M, Dvorak CC, Gehring S, Gennery A, et al. The syndrome of hemophagocytic lymphohistiocytosis in primary immunodeficiencies: implications for differential diagnosis and pathogenesis. Haematologica. 2015;100(7):978-988.
doi pubmed - Rui R, Zhou L, He S. Cancer immunotherapies: advances and bottlenecks. Front Immunol. 2023;14:1212476.
doi pubmed - Filipovich A, McClain K, Grom A. Histiocytic disorders: recent insights into pathophysiology and practical guidelines. Biol Blood Marrow Transplant. 2010;16(1 Suppl):S82-89.
doi pubmed - Daver N, McClain K, Allen CE, Parikh SA, Otrock Z, Rojas-Hernandez C, Blechacz B, et al. A consensus review on malignancy-associated hemophagocytic lymphohistiocytosis in adults. Cancer. 2017;123(17):3229-3240.
doi pubmed - Poto R, Troiani T, Criscuolo G, Marone G, Ciardiello F, Tocchetti CG, Varricchi G. Holistic approach to immune checkpoint inhibitor-related adverse events. Front Immunol. 2022;13:804597.
doi pubmed - Rajapakse P, Andanamala H. Hemophagocytic lymphohistiocytosis secondary to immune checkpoint inhibitor therapy. World J Oncol. 2022;13(2):49-52.
doi pubmed - Brisse E, Wouters CH, Matthys P. Advances in the pathogenesis of primary and secondary haemophagocytic lymphohistiocytosis: differences and similarities. Br J Haematol. 2016;174(2):203-217.
doi pubmed - Canna SW, Marsh RA. Pediatric hemophagocytic lymphohistiocytosis. Blood. 2020;135(16):1332-1343.
doi pubmed - Jordan MB, Allen CE, Weitzman S, Filipovich AH, McClain KL. How I treat hemophagocytic lymphohistiocytosis. Blood. 2011;118(15):4041-4052.
doi pubmed - De Matteis A, Colucci M, Rossi MN, Caiello I, Merli P, Tumino N, Bertaina V, et al. Expansion of CD4dimCD8+ T cells characterizes macrophage activation syndrome and other secondary HLH. Blood. 2022;140(3):262-273.
doi pubmed - Janka GE. Hemophagocytic syndromes. Blood Rev. 2007;21(5):245-253.
doi pubmed - Janka GE, Lehmberg K. Hemophagocytic syndromes—an update. Blood Rev. 2014;28(4):135-142.
doi pubmed - Henter JI, Horne A, Arico M, Egeler RM, Filipovich AH, Imashuku S, Ladisch S, et al. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48(2):124-131.
doi pubmed - Maude SL, Barrett D, Teachey DT, Grupp SA. Managing cytokine release syndrome associated with novel T cell-engaging therapies. Cancer J. 2014;20(2):119-122.
doi pubmed - Naymagon L. Anakinra for the treatment of adult secondary HLH: a retrospective experience. Int J Hematol. 2022;116(6):947-955.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Medical Cases is published by Elmer Press Inc.