| Journal of Medical Cases, ISSN 1923-4155 print, 1923-4163 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Med Cases and Elmer Press Inc |
| Journal website https://jmc.elmerpub.com |
Case Report
Volume 16, Number 3, March 2025, pages 114-119

Medium-Chain Acyl-CoA Dehydrogenase Deficiency Disorder as a Cause of Acute Liver Failure in a 23-Month-Old Baby
Inva Gjetaa, b, Ilirjana Bakallia, Durim Salaa, Ermela Celaja, Marsela Biqikua, Vladimir Hoxhaa, Virtut Velmishia, Elmira Kolaa
aPediatric Intensive Care Unit, University Hospital Center “Mother
Teresa”, Tirana, Albania
bCorresponding Author: Inva Gjeta, Pediatric
Intensive Care Unit, University Hospital Center “Mother Teresa”, Tirana, Albania
Manuscript submitted October 15, 2024, accepted February 18, 2025, published online February 27,
2025
Short title: MCAD Deficiency as a Cause of Acute Liver Failure
doi:
https://doi.org/10.14740/jmc5093
| Abstract | ▴Top |
Fatty acid oxidation disorders are inborn metabolic defects caused by impaired beta-oxidation of fats within the mitochondria. This occurs due to a deficiency in the pathway of fatty acids into the mitochondria via carnitine. Although their incidence is not frequent, the clinical presence of this disorder often leads to morbidity and high mortality. Medium-chain acyl-CoA dehydrogenase (MCAD) deficiency is part of the large group of fatty acid oxidation disorders which has a high variability in clinical manifestations and in daily medical practice can be challenging to early and correctly diagnose. In this article, we present a 23-month-old boy with drowsiness, mild hypoglycemia, and rapid progression to acute liver failure as a consequence of this metabolic disorder. Once the diagnosis was confirmed, treatment was conducted following the guideline of hypoglycemia of the metabolic disorder of MCAD deficiency and its complications. The child was discharged in good condition and the follow-up after 6 months was successful. Further, we review the literature on this genetic condition and check on how they connect to our case. The article aims to focus on the early evaluation of the clinical signs that present from the underlying of this rare metabolic disorder and the importance of aggressive treatment to prevent complications that can be fatal for the patient.
Keywords: Acyl-CoA dehydrogenase; Liver failure; Hypoglycemia; Fatty oxidation
| Introduction | ▴Top |
Medium-chain acyl-CoA dehydrogenase (MCAD) deficiency is one of the most common inherited fatty acid beta-oxidation disorders. Depending on the affected population, the disease has a variable incidence. The reported global incidence ranges from 1:4,000 to 1:15,000 [1, 2]. The highest incidence was found in the populations of northern Europe. In North America, the prevalence varies from 1:24,000 live births in Canada to 1:13,000 to 1:19,000 in other states of the United States [3-6].
The MCAD deficiency disease is transmitted in an autosomal recessive manner and the enzyme deficiency prevents fatty acids from being used as an energy source. During periods of catabolic growth, prolonged fasting, viral infections, physical exertion, the depletion of glucose storage in the liver, the reduction of the process of ketogenesis and the production of ketone bodies bring an imbalance of energy homeostasis in the organism [1, 7-9].
Clinical manifestations associated with this disorder are general weakness, lack of appetite, drowsiness, hepatic dysfunction with increased transaminases up to acute liver failure, coma, and sudden death [1, 7, 8]. In this article, we present a boy with acute hepatic failure. The initial signs were vomiting, drowsiness, mild hypoglycemia and later rapid progression to acute liver damage co-associated by a sub-comatose state. Although liver injury is described as part of the clinical syndrome that comes from MCAD deficiency disease, reports of such cases in the literature are not frequent. In this article, the focus is placed on the early clinical suspicion of acute hepatic encephalopathy as a consequence of metabolic dysregulation and aggressive treatment to inhibit the catabolic process that could be life-threatening for the patient.
| Case Report | ▴Top |
A 23-month-old child presented to the pediatric intensive care unit with frequent vomiting, diarrhea, several episodes, no blood, a temperature of 38.5 °C, and debilitation. According to his parents, the child had been in this state for 2 days, and in the last few hours, the general condition had worsened.
Medical records show a full-term pregnancy and cesarean delivery, and the baby was 4,200 g at birth. No problem had been recorded in the postpartum period. The parents are not related by consanguinity.
According to his parents, at 13 months, the boy was treated for acute gastroenteritis, and due to the few oral intakes, he was treated in the emergency room with parenteral rehydration for several hours. After this episode, the child did not show any problems until the moment of hospitalization.
During hospitalization, the child was quarrelsome and sometimes sleepy. He would wake up completely, be verbally responsive, and have adequate pupillary reflex to light. During the visit, the child looked dehydrated and pale, with sunken and dark circles under the eyes, dry oral mucosa, no conjunctival injection, with decreased skin turgor. Physical examinations showed no sign of nuchal rigidity around the neck, no Bruzhinski’s and Kerning’s signs, except noticeable drowsiness; slightly tachycardic rhythmic tones, no cardiac murmur with a frequency 160/min; clear auscultation lungs, no rhonchi and rales, without dyspnea; soft abdomen, not painful, without hepatomegaly and splenomegaly; no asymmetry or deficit on extremities, without edema, no enlargement of lymph nodes in the axillary and inguinal areas; normal genital examination.
Laboratory test revealed complete blood count with red blood cells (RBCs) of 3.96 × 106/mm3 (range: 4.2 - 6.1 × 106/mm3), white blood cells (WBCs) of 13.2 × 103/mm3 (range 4 - 12 × 103/mm3), hemoglobin (Hb) level of 10.9 g/dL (range: 1 - 16 g/dL), and platelet (PLT) of 307 × 103/mm3 (range: 150 - 390 × 103).
Biochemical test results showed blood glucose of 60 mg/dL (range: 70 - 120 mg/dL), urea nitrogen of 37.2 mg/dL (range: 10 - 43 mg/dL), creatinine of 0.5 mg/dL (range: 0.6 - 1.2 mg/dL), gamma-glutamyl transferase (GGT) of 4 U/L (range: 12 - 64 U/L), aspartate aminotransferase (AST) of 40 U/L (range: 0 - 35 U/L), alanine transaminase (ALT) of 19 U/L (range: 0 - 45 U/L), lactate dehydrogenase (LDH) of 291 U/L (range: 125 - 250 U/L), total bilirubin of 0.72 mg/dL (range: 0.3 - 1.2 mg/dL), direct bilirubin of 0.34 mg/dL (range: 0.1 - 0.3 mg/dL), total protein of 4.5 g/dL (range: 6 - 8.3 g/dL), albumin of 3.2 mg/dL (range: 3.4 - 5.2 g/dL), Na of 136 mEq/L (range: 135 - 145 mEq/L), K of 3.9 mEq/L (range: 3.5 - 5.1 mEq/L), C-reactive protein of 0.89 mg/dL (range: 0 - 0.5 mg/dL), prothrombin time activity (PTA) of 76% (range: 70-120%) and international normalized ratio (INR) of 1.1 (range: 0.7 - 1.2).
Urine test showed normal urine, no leukocytes, normal pH, and no glucose, with traces of ketones present. Hemoculture and urine culture were taken.
Blood gas analysis results showed a pH of 7.34, HCO3 of 15.8 mmol/L, and base excess in extracellular fluid (BEecf) of -13 mmol/L. Abdominal ultrasound was normal. Chest X-ray was normal.
Initially, the child was treated for acute moderate isotonic dehydration in the context of a gastrointestinal viral infection. A bolus was performed with 25% glucose in a dose of 2 mL/kg/weight for the hypoglycemia of 60 mg/dL. The dehydration scale by percentage was evaluated to be 6-7%, and the treatment was done according to the daily requirement of fluids and the deficit, with saline solution and 5% glucose in the ratio 1:1 and electrolytes. Blood glucose measurements during the 16 h of hospitalization were normal.
The following day, the child’s condition did not improve; however, he did not seem dehydrated. The sleepiness persisted, and episodes of awakening were rarer. The temperature persisted up to 38.5 °C, with fewer episodes of diarrhea. Normal diuresis was present.
An acute meningitis was ruled out through the examination of the cerebral liquor with a normal result of three cells in cerebrospinal liquid. Glucose and protein were normal as well in the cerebrospinal liquor. Real-time transcriptase-polymerase chain reaction (RT-PCR) for severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) was negative.
Biochemical test results of the next day showed glycemia of 75 mg/dL (range: 70 - 120 mg/dL), total protein of 4.3 g/dL (range: 6 - 8.3 g/dL), albumin of 2.9 mg/dL (range: 3.4 - 5.2 g/dL), ALT of 29 U/L (range: 0 - 45 U/L), AST of 55 U/L (range: 0 - 35 U/L), LDH of 337 U/L (range: 125 - 250 U/L), creatine kinase (CK) level of 383 U/L (range: 30 - 170 U/L), and blood ammonia level of 339 µg/dL (range: 40 - 80 µg/dL). The results of glycemia measurement and ammonia level in blood during the days are shown in Table 1.
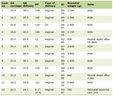 Click to view |
Table 1. Blood Glucose and Ammonia
Levels |
In the objective examination, there was an increase in the liver palpation of 3 cm. As the clinical conditions did not improve with the persistent doziness, and non-response of rehydration therapy, excluding an acute infection such as meningoencephalitis and high level of ammonia, then the diagnostic approach was directed into hepatic encephalopathy of an unclear nature.
Even though the patient was treated with parenteral therapy with 5% glucose and saline solution, in the next examination, there were still traces of ketones, and the blood glucose test during the last 24 h showed three different measurements of hypoglycemia. The abdominal ultrasound on the second day showed an increase in the liver size at about 11 cm and a rough structure of it.
In the presence of encephalopathy, hepatomegaly, and hypoglycemia, a metabolic disorder was suspected, and we started with the analysis of acetylcarnitine profile in the plasma.
The child was treated with 10% glucose IV at a rate of 1.5 times a day as needed, electrolytes, and bicarbonate salt. Subsequent analyses confirmed an acute hepatic failure with ALT of 644 U/L (range: 0 - 45 U/L), AST of 1,052 U/L (range: 0 - 35 U/L), CK level of 3,117 U/L (range: 125 - 250 U/L), LDH of 1,708 U/L (range: 125 - 250 U/L), PTA of 25% (range: 70-120%), and INR of 2.87 (range: 0.7 - 1.2).
After aggressive treatment with 10% glucose infusion, electrolytes, human albumin 1 g/kg/weight per day, vitamin K, and plasma transfusion, the child’s condition improved significantly.
The analysis results of the acetylcarnitine profile were high: hexanoylcarnitine (C6) was 0.85 µmol/L with a normal range of 0.01 - 0.22 µmol/L, octanoylcarnitine (C8) was 4.46 µmol/L with a normal range of 0.01 - 0.44 µmol/L.
Genetic analyses through exome sequencing confirmed the MCAD deficiency disease. The responsible variant in ACADM was C.244dup.p9 (Trp82Leufs15).
The child’s condition improved and a follow-up by a metabolic specialist was recommended regarding the diet plan and the following preventive measures. The 6-month follow-up showed a child in good health.
Genetic counseling for the parents was recommended.
| Discussion | ▴Top |
MCAD deficiency is a genetic condition that results from an enzymatic deficit or reduced activity of the acyl-CoA dehydrogenase enzyme. Enzymes belong to the family of flavoproteins [10]. The gene responsible for the disease is ACADM, which is located on chromosome 1p31 and enables the coding of the enzymatic protein acetyl-CoA dehydrogenase [11]. This enzyme is needed as a catalyst for the process of oxidation of fatty acids in the mitochondria, serving as the first step of the fatty acid oxidation cycle and later metabolizes the production of energy in the form of adenosine triphosphate (ATP) and ketone bodies [12, 13].
Mutations in deoxyribonucleic acid (DNA) that replace guanine with adenine at 985 position of the coding part have been observed in 90% of cases of mutant genes in MCAD deficiency disorder [14]. Today, there are about 80 allelic variants reported in different literature [13].
This disorder is transmitted in an autosomal recessive manner and presents itself with a variable incidence depending on the affected ethnic population. The total prevalence of MCAD deficiency disease is calculated at 5.3 per 100,000 live births in different populations [6]. In a healthy organism, during periods of stress, when energy demands increase, energy homeostasis after the consumption of glucose stored in the liver is realized through the process of lipolysis [1]. In MCAD deficiency disease, this process is altered, bringing the deposition of fatty acids in the liver, with reduced ketones in blood or absence, and with persistent hypoglycemia. The clinical signs that prevail in these patients include weakness, drowsiness, lethargy, and seizures as a result of hypoglycemia [8]. It is also noted the presence of hepatic dysfunction with hepatomegaly, sub-comatose state, acute hepatic encephalopathy, Reye-like syndrome as a result of increased transaminases, infiltration of fatty acids in the liver, and increased level of ammonia leading to cerebral edema [8, 15].
The MCAD deficiency disorder can be diagnosed at any age, newborn, early childhood, adolescent, and adult, but the most frequent reports have been identified in the neonatal age up to 2 years old [7, 8, 16, 17].
Individuals affected by MCAD deficiency disorder present a great diversity in the clinical spectrum, which is influenced by several factors, such as the age at which time the symptoms appear, the inciting circumstances or trigger factors, and the individual features of the genotype-phenotype relationship, influencing the severity of the disease [8]. These factors, being influenced by each other, make this disease very heterogeneous and multiplanar in its presentation [8, 18, 19].
In the neonatal age, this disease can appear with the syndrome of sudden death and often remains undiagnosed [20-22]. Sudden death has been reported in adults as well [16].
In the first phase of the diagnostic approach, the probable presence of a viral infection, negative toxidrome in the history, traces of ketones in the urine, and the rare incidence of metabolic disorders suggested a hepatic insufficiency unknown, probably in the context of Reye syndrome. Although the history was negative in the use of aspirin, it could serve as a trigger. Then the hypoglycemia that was recorded during the blood glucose measurements and the brief deterioration in the level of consciousness made us suspect a metabolic disorder.
In different literature, it has been noticed that in MCAD deficiency disease, we do not always have an absolute lack of urine ketones [8]. The presence of traces of ketones in urine in the phases of metabolic crisis as a result of dehydration and oxidation of long-chain fatty acids often serves as a “trap” for delay in diagnosis in clinical practice [8, 23].
In our patient, the elements that helped in the diagnosis were persistent hypoglycemia, although not severe, and the rapid precipitation of the clinical condition, from a child initially in a stable state, without hepatic damage to progressing to a sudden onset of acute hepatic failure with coma.
In patients with MCAD deficiency, in addition to the liver, which remains the most affected organ, neurological manifestations have also been observed, such as reduced neurocognitive development ability or attention-deficit hyperactivity disorder (ADHD) [8, 24]. Muscular pain, chronic fatigue, and intolerance to physical exertion are part of the clinical diversity of MCAD deficiency disease as well [8]. Further, cardiac involvement in MCAD deficiency disease is reported, although in rare cases, in the form of rhythm disorders and prolongation of the QT interval [8]. Renal manifestations are in the form of chronic renal disease as a result of the infiltration of toxic sebaceous metabolites in the renal tubules [8].
The diagnosis of MCAD deficiency disease is often difficult, with a wide age range and not infrequently with signs that are not clear or suspicious, but which can be perceived quickly, endangering the patient’s life [8]. The diagnosis is based on clinical data and confirmatory tests.
Neonatal screening is one of the tests performed directly after birth. The evaluation is done through tandem mass spectrometry on dried blood spots where the level of medium-chain acetyl carnitines and methods chosen for the diagnosis of MCAD deficiency require an interpretation correlated with the clinical indicators [8, 24-27]. C6 and C10 are also high and C8/C10 has been found 10 times higher in patients with MCAD deficiency, serving with value in the diagnosis of these individuals [8, 28].
The level of organic acids in urine is a test that has value in symptomatic individuals with MCAD deficiency [29]. In the urine of patients with MCAD deficiency, we have a high level of medium-chain dicarboxylic acids with a trend indicator. The test is based on measuring the high level of C8-acylcarnitine through the liquid chromatography-tandem mass spectrometry method in the blood [8, 24-27]. C6 and C10 levels are also high and C8/C2 and C8/C10 ratios are also high. In the literature, level of dicarboxylic acids follow up these trends hexanonylglycine (6) > octanoylglycine (C8) > decanoylglycine (C10). The level of ketones in the urine is absent or very low. It should be considered that the lack of ketones is not a specific sign in the diagnosis of MCAD deficiency, since in the course of metabolic crises, we have its presence in the urine, which can also lead to misdiagnosis [1, 8].
Analysis of arylglycine in urine determines high levels of n-hexanoyl glycine, 3-propionylglycine, and suberylglycine. This test is more informative in cases of asymptomatic patients or with mild symptoms [1, 8, 29, 30].
DNA genetic tests will be used depending on the clinical signs or the results of the previous tests. Depending on the clinical signs, the history of the disease, and the family history of the patient, single targeting gene, multigene panel, and exome sequencing or whole genome can be used for the most difficult diagnostic cases [1, 24].
Enzymatic activity measurement of the MCAD enzyme is used mainly in cases where we have mutant variants without significance in genetic tests or the presence of a pathogenic variant [31]. The test consists in measuring the enzymatic activity of MCAD in the culture of fibroblasts or other tissues such as leukocytes, liver, muscles, etc. [8, 32]. Patients will be considered with MCAD deficiency with enzyme activity < 10% [32]. In another study, the enzymatic activity reaches < 35% in patients with MCAD deficiency [33, 34].
Treatment of MCAD deficiency disease
The treatment does not include a specific cure for the disease. There are two main lines in the treatment of the acute phase and the prevention of clinical signs and management of complications.
Acute management focuses on the treatment of hypoglycemia and inhibition of the catabolic process that accompanies acute decompensation and the evaluation and treatment of the factors that precipitate the disease [8, 35].
The treatment aims to give liquids that contain amounts of carbohydrates or parenteral glucose depending on the patient’s symptoms. Metabolic crisis in patients with MCAD deficiency should be considered a medical emergency, as a delay in treatment has a high risk of fatal progression of the disease [35, 36]. Hypoglycemia is treated with solution dextrose 25% IV bolus at a dose of 2 mL/kg/weight [35, 36].
For patients who do not tolerate oral treatments, who have vomiting, loss of consciousness, signs of dehydration or documented hypoglycemia even when tolerated orally, it is recommended to start therapy with dextrose infusion 10% IV with a dose of 1.5 times the daily requirement or 10 - 12 mg/kg/min glucose to maintain blood glucose level above 5 mmol/L or between 120 and 170 mg/dL [8, 15, 35, 36].
Prevention of clinical signs aims to avoid prolonged fasting. Nutrition guidelines by genetic metabolic dieticians recommend a fasting time of 4 h for children under 4 months, with an additional hour for each month of age up to 12 months. For children 12 - 24 months up to 10 h, and for children over 24 months fasting goes to 12 h [8, 37]. Before going to bed, it is recommended to feed the child with 2 g/kg/weight of uncooked cornstarch to maintain a linear level of glucose during sleep [8, 35]. A healthy, balanced diet is also recommended, not limiting fats, but the calories from these should not exceed > 30% of the total energy [8]. Physical effort or variable sports should be supported with carbohydrates and rehydration therapy. Patients are advised to avoid foods containing medium-chain triglycerides, coconut oil, diets that include prolonged fasting, not using alcohol in teenagers and aspirin [8, 17].
In different literature, supplements with carnitine have been found contradictory because of increased renal excretion of carnitine in patients with MCAD deficiency, leading to a secondary deficit of it. Its use for correcting the deficit and improving increased tolerance to physical effort is recommended by many researchers [8, 38, 39]. On the other hand, long-term use brings side effects, and it has not been found to improve fasting [8]. However, its use in low doses and in patients with low levels of carnitine, in patients with severe forms of MCAD deficiency intolerant to physical exertion and marked muscle weakness finds support from many authors [8, 40, 41].
The innovative therapy is described as the therapy with glycerol phenylbutyrate, as an adjuvant and is still in the clinical trial phase in adults [8, 24]. Gene therapy with adenovirus vector has shown promising data, but the studies are still in animals [24].
Genetic counseling is needed, and it is necessary that the patient has information about the disease, its transmission, the chances of how it affects the future of the family, and the quality of life of the affected persons [8].
Conclusions
MCAD deficiency is one of the most frequent disorders of fatty beta-oxidation and the clinical signs are often complex with a wide large presentation. The early recognition of it is important for clinical decisions and affects the outcome. Early and aggressive treatment is lifesaving for patients with MCAD deficiency.
Learning points
MCAD deficiency can present as an acute liver failure in medical practice. Red flag signs like hypoglycemia should be taken into consideration for a careful diagnostic approach for a correct diagnosis.
Acknowledgments
None to declare.
Financial Disclosure
None to declare.
Conflict of Interest
None to declare.
Informed Consent
Informed consent has been obtained.
Author Contributions
IGJ, IB and EK conceptualized the idea for this article and edited the final manuscript. EC, MB and VV helped in writing the paper and collect the information. VH and DS supervised the patient care.
Data Availability
The authors declare that data supporting the findings of this study are available within article.
Abbreviations
ADHD: attention-deficit hyperactivity disorder; MCAD: medium-chain acyl-CoA dehydrogenase; RT-PCR: real-time transcriptase-polymerase chain reaction; SARS-CoV-2: severe acute respiratory syndrome coronavirus 2
| References | ▴Top |
- Merritt JL, 2nd, MacLeod E, Jurecka A, Hainline B. Clinical
manifestations and management of fatty acid oxidation disorders. Rev Endocr Metab Disord.
2020;21(4):479-493.
doi pubmed pmc - Lindner M, Hoffmann GF, Matern D. Newborn screening for
disorders of fatty-acid oxidation: experience and recommendations from an expert meeting.
J Inherit Metab Dis. 2010;33(5):521-526.
doi pubmed - Prasad C, Speechley KN, Dyack S, Rupar CA, Chakraborty P,
Kronick JB. Incidence of medium-chain acyl-CoA dehydrogenase deficiency in Canada using the
Canadian Paediatric Surveillance Program: role of newborn screening. Paediatr Child Health.
2012;17(4):185-189.
doi pubmed pmc - Chace DH, Kalas TA, Naylor EW. The application of tandem
mass spectrometry to neonatal screening for inherited disorders of intermediary metabolism. Annu
Rev Genomics Hum Genet. 2002;3:17-45.
doi pubmed - Frazier DM, Millington DS, McCandless SE, Koeberl DD, Weavil
SD, Chaing SH, Muenzer J. The tandem mass spectrometry newborn screening experience in North
Carolina: 1997-2005. J Inherit Metab Dis. 2006;29(1):76-85.
doi pubmed - Feuchtbaum L, Carter J, Dowray S, Currier RJ, Lorey F. Birth
prevalence of disorders detectable through newborn screening by race/ethnicity. Genet Med.
2012;14(11):937-945.
doi pubmed - Medium chain acyl-CoA dehydrogenase deficiency. Cincinnati Children’s. https://www.cincinnatichildrens.org/search?q=Medium%20chain%20acyl-Coa%20dehydrogenase%20deficiency.
- Chang IJ, Lam C, Vockley J. Medium-chain acyl-coenzyme A
dehydrogenase deficiency. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A,
editors. GeneReviews®. Seattle (WA): University of Washington; 1993. Last
updated 2019.
pubmed - Vockley J, Hahn S, Kremen J. Overview of fatty acid oxidation disorder. UpToDate. 2024. https://www.uptodate.com/contents/overview-of-fatty-acid-oxidation-disorders.
- Crane FL, Hauge JG, Beinert H. Flavoproteins involved in the
first oxidative step of the fatty acid cycle. Biochim Biophys Acta. 1955;17(2):292-294.
doi pubmed - Morris AM, Spiekerkoetter U. Disorder of mitochondrial fatty acid oxidation and related metabolic pathways. In: Saudubray JM, van den Berghe G, Walter JH, editors. Inborn metabolic disease: diagnosis and treatment. New York: Springer; 2012. p. 201-216.
- Leydiker KB, Neidich JA, Lorey F, Barr EM, Puckett RL, Lobo
RM, Abdenur JE. Maternal medium-chain acyl-CoA dehydrogenase deficiency identified by newborn
screening. Mol Genet Metab. 2011;103(1):92-95.
doi pubmed - Roth KS. Medium chain acyl-CoA dehydrogenase deficiency. Medscape. 2021. https://emedicine.medscape.com/article/946755-overview?_gl=1*1ddg91m*_gcl_au*MTg3NDg4NTU4OC4xNzQwMjgxNTg5&form=fpf.
- Wilcken B, Hammond J, Silink M. Morbidity and mortality in
medium chain acyl coenzyme A dehydrogenase deficiency. Arch Dis Child.
1994;70(5):410-412.
doi pubmed pmc - Saudubray JM, Martin D, de Lonlay P, Touati G, Poggi-Travert
F, Bonnet D, Jouvet P, et al. Recognition and management of fatty acid oxidation defects: a
series of 107 patients. J Inherit Metab Dis. 1999;22(4):488-502.
doi pubmed - Raymond K, Bale AE, Barnes CA, Rinaldo P. Medium-chain
acyl-CoA dehydrogenase deficiency: sudden and unexpected death of a 45 year old woman. Genet
Med. 1999;1(6):293-294.
doi pubmed - Lang TF. Adult presentations of medium-chain acyl-CoA
dehydrogenase deficiency (MCADD). J Inherit Metab Dis. 2009;32(6):675-683.
doi pubmed - Touw CM, Smit GP, de Vries M, de Klerk JB, Bosch AM, Visser
G, Mulder MF, et al. Risk stratification by residual enzyme activity after newborn screening for
medium-chain acyl-CoA dehydrogenase deficiency: data from a cohort study.
Orphanet J Rare Dis. 2012;7:30.
doi pubmed pmc - Gartner V, McGuire PJ, Lee PR. Child neurology: medium-chain
acyl-coenzyme A dehydrogenase deficiency. Neurology. 2015;85(4):e37-e40.
doi pubmed pmc - Iafolla AK, Thompson RJ, Jr., Roe CR. Medium-chain
acyl-coenzyme A dehydrogenase deficiency: clinical course in 120 affected children.
J Pediatr. 1994;124(3):409-415.
doi pubmed - Rinaldo P, Matern D, Bennett MJ. Fatty acid oxidation
disorders. Annu Rev Physiol. 2002;64:477-502.
doi pubmed - Alonso EM. Acute liver failure in children: the role of
defects in fatty acid oxidation. Hepatology. 2005;41(4):696-699.
doi pubmed - Maduemem KE. Medium-chain acyl-coenzyme A dehydrogenase
deficiency (MCADD): a cause of severe hypoglycaemia in an apparently well child. BMJ Case Rep.
2016;2016:bcr2016217538.
doi pubmed pmc - Ibrahim SY, Vaqar S, Temtem T. Medium-chain acyl-CoA
dehydrogenase deficiency. In: StatPearls. Treasure Island (FL): StatPearls Publishing;
2025.
pubmed - Minkler PE, Stoll MSK, Ingalls ST, Hoppel CL. Correcting
false positive medium-chain acyl-CoA dehydrogenase deficiency results from newborn screening;
synthesis, purification, and standardization of branched-chain C8 acylcarnitines for use in
their selective and accurate absolute quantitation by UHPLC-MS/MS. Mol Genet Metab.
2017;120(4):363-369.
doi pubmed - Millington DS, Kodo N, Norwood DL, Roe CR. Tandem mass
spectrometry: a new method for acylcarnitine profiling with potential for neonatal screening for
inborn errors of metabolism. J Inherit Metab Dis. 1990;13(3):321-324.
doi pubmed - Niwa T. Mass spectrometry in disorders of organic acid
metabolism. Clin Chim Acta. 1995;241-242:293-384.
pubmed - Tucci S, Wagner C, Grunert SC, Matysiak U, Weinhold N, Klein
J, Porta F, et al. Genotype and residual enzyme activity in medium-chain acyl-CoA dehydrogenase
(MCAD) deficiency: are predictions possible? J Inherit Metab Dis.
2021;44(4):916-925.
doi pubmed - Rinaldo P, Studinski AL, Matern D. Prenatal diagnosis of
disorders of fatty acid transport and mitochondrial oxidation. Prenat Diagn.
2001;21(1):52-54.
doi pubmed - Rinaldo P, Raymond K, al-Odaib A, Bennett MJ. Clinical and
biochemical features of fatty acid oxidation disorders. Curr Opin Pediatr.
1998;10(6):615-621.
doi pubmed - Alcaide P, Ferrer-Lopez I, Gutierrez L, Leal F,
Martin-Hernandez E, Quijada-Fraile P, Bellusci M, et al. Lymphocyte medium-chain acyl-CoA
dehydrogenase activity and its potential as a diagnostic confirmation tool in newborn screening
cases. J Clin Med. 2022;11(10):2933.
doi pubmed pmc - Hale DE, Stanley CA, Coates PM. Genetic defects of acyl-CoA
dehydrogenases: studies using an electron transfer flavoprotein reduction assay. Prog Clin Biol
Res. 1990;321:333-348.
pubmed - Bouvier D, Vianey-Saban C, Ruet S, Acquaviva C. Development
of a tandem mass spectrometry method for rapid measurement of mmedium- and very-long-chain
acyl-CoA dehydrogenase activity in fibroblasts. JIMD Rep. 2017;35:71-78.
doi pubmed pmc - Mason E, Hindmarch CCT, Dunham-Snary KJ. Medium-chain
acyl-CoA dehydrogenase deficiency: pathogenesis, diagnosis, and treatment. Endocrinol Diabetes
Metab. 2023;6(1):e385.
doi pubmed pmc - McGregor TL, Berry SA, Dipple KM, Hamid R, Council on
Genetics Collaborators. Management principles for acute illness in patients with medium-chain
acyl-coenzyme A dehydrogenase deficiency. Pediatrics. 2021;147(1):e2020040303.
doi pubmed - New England Consortium of Metabolic Programs. Medium chain acyl-CoA dehydrogenase deficiency. https://www.newenglandconsortium.org/an-educators-guide-to-mcadd.
- Derks TG, Reijngoud DJ, Waterham HR, Gerver WJ, van den Berg
MP, Sauer PJ, Smit GP. The natural history of medium-chain acyl CoA dehydrogenase deficiency in
the Netherlands: clinical presentation and outcome. J Pediatr.
2006;148(5):665-670.
doi pubmed - Roe CR, Ding J. Mitochondrial fatty acid oxidation disorders. In: Scriver CR, Beaudet AL, Sly WS, Valle D, Childs B, Kinzler KW, Wogelstein B, editors. The metabolic and molecular bases of inherited disease. New York, NY: McGraw-Hill; 2001. p. 2297-2326.
- Lee PJ, Harrison EL, Jones MG, Jones S, Leonard JV, Chalmers
RA. L-carnitine and exercise tolerance in medium-chain acyl-coenzyme A dehydrogenase (MCAD)
deficiency: a pilot study. J Inherit Metab Dis. 2005;28(2):141-152.
doi pubmed - Jager EA, Schaafsma M, van der Klauw MM, Heiner-Fokkema MR,
Derks TGJ. Plasma carnitine concentrations in medium-chain acyl-CoA dehydrogenase deficiency:
lessons from an observational cohort study. J Inherit Metab Dis.
2022;45(6):1118-1129.
doi pubmed pmc - Rucklova K, Hruba E, Pavlikova M, Hanak P, Farolfi M,
Chrastina P, Vlaskova H, et al. Impact of newborn screening and early dietary management on
clinical outcome of patients with long chain 3-hydroxyacyl-CoA dehydrogenase deficiency and
medium chain acyl-CoA dehydrogenase deficiency - a retrospective nationwide study. Nutrients.
2021;13(9):2925.
doi pubmed pmc
This
article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0
International License, which permits unrestricted non-commercial use, distribution, and
reproduction in any medium, provided the original work is properly cited.
Journal
of Medical Cases is published by Elmer Press Inc.