| Journal of Medical Cases, ISSN 1923-4155 print, 1923-4163 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Med Cases and Elmer Press Inc |
| Journal website https://jmc.elmerpub.com/ |
Case Report
Volume 15, Number 12, December 2024, pages 401-405

Gastrointestinal Bleeding/Angiodysplasia in Patients With Glanzmann Thrombasthenia
Raghad A. Tarawaha, c, Ahmad M. Tarawahb
aDepartment of Medicine, King Fahad Hospital, Madinah, Saudi
Arabia
bMadinah Hereditary Blood Disorders Centre, Department of Hematology and
Oncology, King Salman Bin Abdulaziz Medical City, Madinah, Saudi
Arabia
cCorresponding Author: Raghad A. Tarawah, Department of Medicine, King
Fahad Hospital, Madinah, Saudi Arabia
Manuscript submitted September 23, 2024, accepted October 18, 2024, published online November 11,
2024
Short title: GI Angiodysplasia in Glanzmann Thrombasthenia
doi:
https://doi.org/10.14740/jmc4340
| Abstract | ▴Top |
Glanzmann thrombasthenia (GT) is a common type of bleeding disorder, with a prevalence of 1/10,000 in Al Madinah, Saudi Arabia. GT causes bleeding owing to the lack of platelet aggregation associated with glycoprotein IIb/IIIa deficiency, which is characterized by mucocutaneous bleeding symptoms, such as epistaxis, gingival bleeding, and menorrhagia. Gastrointestinal angiodysplasia (GIAD) is a rare presentation of GT, where eight cases have been reported. GIAD is a vascular malformation of the digestive system caused by abnormal angiogenesis. Treatment of GIAD include surgical resection, electrocoagulation, embolization, and medical therapy with octreotide, thalidomide, and bevacizumab. GIAD has a high tendency to recur. We report the cases of eight patients of different ages who were diagnosed with GT and presented with gastrointestinal bleeding.
Keywords: Gastrointestinal angiodysplasia; Gastrointestinal bleeding; Glanzmann thrombasthenia
| Introduction | ▴Top |
Glanzmann thrombasthenia (GT) is a rare hereditary bleeding disorder. It was first described by Dr. Edward Glanzmann in 1918 and named “hereditare hamorrhagische thrombasthenie”. It is an autosomal recessive disorder, characterized by qualitative or quantitative abnormalities associated with glycoprotein IIb/IIIa deficiency. This protein ensures platelet aggregation and thrombus formation at the injury site. It is characterized by normal platelet counts, but deficient clot retraction and platelet aggregation [1]. GT is a rare disorder with a global incidence of 1/1,000,000. However, this is also prevalent in populations where consanguinity is common. In Saudi Arabia, the prevalence is 1/200,000 [2]. The rate is higher in Madinah, Saudi Arabia, accounting for 1/10,000 [3]. GT presents with a wide range of mucocutaneous bleeding symptoms, such as epistaxis, gum bleeding, menorrhagia, subcutaneous bruises, and bleeding at the circumcision site [4]. The treatments used to manage bleeding episodes include local treatment, platelet transfusion, antifibrinolytic therapy, and recombinant factor VIIa (rFVIIa) therapy [5].
Although gastrointestinal (GI) bleeding is uncommon in these patients, its treatment and diagnosis can be challenging. Gastrointestinal angiodysplasia (GIAD) is a vascular malformation due to abnormal angiogenesis localized in any part of the digestive tract. It is the most common cause of obscure GI bleeding and is usually observed in elderly patients aged 60 years or older due to vascular degenerative changes [6]. Endoscopic intervention is the therapy of choice for GIAD; however, the rate of failure is relatively high, as angiodysplastic lesions can develop in certain areas of the GI tract that are inaccessible by endoscopy [7]. GIAD tends to recur, thus requiring frequent hospitalization and increased transfusion requirements. GIAD rarely occurs in patients with GT and has been reported in a few patients worldwide, most of whom are older adults [6-13]. Herein, we report seven adult patients and one child with GT who presented with GI bleeding, five of whom were diagnosed with GIAD.
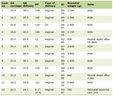
Patients’ information was obtained from the medical record system of the Madinah Hereditary Blood Disorders Center, Maternity and Children Hospital, King Fahd Hospital, Al Madinah, Saudi Arabia. All patients were diagnosed with GT based on the results of flowcytometry and molecular sequencing (Table 1). Of the 130 patients with GT, eight patients presented with upper GI bleeding. Five of them were diagnosed with angiodysplasia by endoscopy, one died prior to further investigation of the cause of bleeding, and two presented with GI bleeding; however, endoscopy was not conclusive. Hence, the patients were treated presumptively for angiodysplasia. Endoscopy was required in those patients as they had severe bleeding not responding to the initial treatment of GT. Table 2 summarizes the main clinical characteristics of all patients.
 Click to view |
Table 1. Laboratory Investigation for GT of the
Eight Cases |
Click to view |
Table 2. Summary of the Main Clinical
Characteristics |
| Case Reports | ▴Top |
Case 1
An 11-year-old girl was known to have type 1 GT, and she was presented by the age of 2 years old. She had no family history of GT. Diagnosis was made using platelet function analyzer-100 (PFA-100) assay, flowcytometry, and molecular sequencing (Table 1). The patient frequently experienced epistaxis and gingival bleeding. Chronic synovitis occurred after hemarthrosis of the right ankle joint. Chemical synovectomy was performed. Severe bleeding from different body sites occurred approximately one to two times per month. Iron deficiency anemia (IDA) was noted. Hence, oral and intravenous (IV) iron supplements were administered. Bone marrow transplantation was recommended, but the patient refused.
The patient presented with melena without vomiting, epistaxis, or evidence of upper GI or fresh rectal bleeding. Recombinant activated factor VIIa (rFVIIa) concentrates were initiated, but no improvement was observed in the patient’s clinical status; subsequently, streaks of fresh blood were observed during defecation. rFVIIa therapy was continued, and platelets were transfused once daily at initial treatment and then increased every 12 h. Melena and hematochezia persisted for 7 days; eventually, hematochezia was resolved after 7 days, while melena was resolved after 10 days. She required the packed red blood cell transfusion twice. Meanwhile, rFVIIa and platelet transfusions were gradually discontinued. The results of the first endoscopy performed during the bleeding episode were inconclusive. Repeated endoscopy in the absence of bleeding showed a stable intestinal angiodysplasia.
Case 2
At the age of 13 years, this female patient with type 1 GT experienced an episode of melena, which was controlled for 5 days with platelet transfusion. Upper GI endoscopy revealed angiodysplasia of the jejunum. At the age of 26 years, the patient experienced recurrent episodes of melena (almost twice a year) and an episode of hematemesis. The patient was treated with recombinant FVIIa and/or a platelet transfusion. Colonoscopy and barium enema examination showed negative results, endoscopy in more than one occasion showed active gastric and jejunum angiodysplasia, and GI angiography confirmed the presence of angiodysplasia. An endoscopic intervention trial was done, but the patient still had bleeding. Endovascular angiography intervention service was not available at the time of her presentation. Surgical resection was not indicated in accordance with the surgeon’s opinion, and octreotide, thalidomide, and bevacizumab treatment were not available.
The patient was diagnosed using PFA-100 assay, flow cytometry, and molecular sequencing (Table 1). She experienced severe bleeding, including menorrhagia, epistaxis, and bruises, once a month.
Case 3
This 46-year-old male patient was diagnosed with type 1 GT by PFA-100 assay, flow cytometry, and molecular sequencing (Table 1). He had siblings with similar conditions. He experienced moderate bleeding (one to two times a year) with IDA, thus requiring long-term IV iron treatment. He initially presented with melena and then eventually experienced vomiting with coffee ground content. After receiving rFVIIa therapy, platelet transfusion, and blood transfusion, the bleeding stopped after 10 days. Multiple angiodysplasias in the duodenum were identified on endoscopic examination. The patient experienced multiple recurrent episodes of upper GI bleeding that were managed in a similar manner.
Case 4
A 39-year-old female patient was diagnosed with type 1 GT using PFA-100 assay, flow cytometry, and molecular sequencing (Table 1). She had a family history of GT. She experienced moderate bleeding approximately one to two times per year in the form of epistaxis, menorrhagia, and hematuria. She also had IDA and received oral and IV iron supplements. At 20 years of age, she experienced upper GI bleeding with melena. Hence, blood transfusion, platelet transfusion, and rFVIIa therapy were administered. Repeat endoscopy revealed the presence of a single angiodysplasia.
Case 5
A 48-year-old male patient was diagnosed with type 1 GT using PFA-100 assay, flow cytometry, and molecular sequencing (Table 1). His two siblings and five of his children were previously diagnosed with GT. The patient experienced moderately severe bleeding with epistaxis, occurring approximately one to two times per year. At the age of 39 years, he experienced an episode of hematemesis and developed melena that persisted for 3 weeks, which were managed by rFVIIa therapy and platelet transfusion; the bleeding eventually stopped. Endoscopic examination revealed the presence of duodenal angiodysplasia.
Case 6
An 18-year-old male patient with an uncle and two other siblings who were previously diagnosed with GT type 1 experienced mild episodes of bleeding approximately once a year, with gum bleeding, bruises, and ecchymosis. He presented with melena and a few episodes of hematochezia; the hemoglobin (Hb) level was 10.8 upon admission. The patient’s bleeding persisted for 17 days, and the Hb level significantly reduced to 5.3. During hospital admission, multiple blood transfusions, platelet transfusion, and rFVIIa therapy were administered. In the last 2 days, he experienced severe intractable fresh rectal bleeding and eventually died. An endoscopic examination or other investigations could not be performed owing to the patient’s general condition.
Cases 7, 8
Two brothers aged 42 and 35 years, with type 1 GT, experienced mild episodes of bleeding; both had IDA requiring long-term oral and IV iron supplementation. They experienced upper GI bleeding with melena and hematemesis for 4 - 5 days. An endoscopy was performed in both patients, but no abnormalities were observed. They were treated presumptively for angiodysplasia with blood transfusion, platelet transfusion, rFVIIa, which successfully stopped the bleeding.
| Discussion | ▴Top |
GIAD is the most common cause of occult GI bleeding in older adults. Its incidence increases in patients with end-stage renal disease, von Willebrand disease, and aortic stenosis; however, the underlying mechanism is not well understood [6]. GIAD is usually diagnosed by endoscopy; in some cases, diagnosis is made based on the findings of radiographic imaging or surgery. GIAD can be managed using a variety of endoscopic interventions, including cautery, argon plasma coagulation, and endoscopic clips; however, as GIAD tends to recur after endoscopic procedures, other modalities can be used, such as angiography with embolization or surgery [14]. Recently, new medications have been used to treat refractory angiodysplasia including anti-vascular endothelial growth factor agents such as thalidomide and bevacizumab [6]. Duarte et al [6] and Paciullo et al [8] reported that two elderly patients with GT experienced recurrent GI bleeding due to GIAD, which was managed by thalidomide; GI bleeding was controlled. Hence, hospitalization, platelet, and blood transfusion were reduced. However, one of the patients died from GI bleeding and sepsis after 6 months. Two other GT patients with GIAD reported by Marlu et al [7] and Barre et al [9] experienced bleeding that was refractory to other treatment options, such as endoscopic intervention, octreotide, and hormonal therapy. Bevacizumab therapy was initiated, and the bleeding episodes were eventually resolved. Furthermore, octreotide combined with hormonal therapy (ethinylestradiol + norethisterone pill) has been suggested by Coppola et al [10] in GT patients with GI bleeding; however, bleeding did not improve, but the transfusion requirement was decreased. Leach et al [11] also described the use of progesterone-only pills to reduce transfusion requirements in a GT middle-aged woman with GIAD.
A recent international prospective register described the treatment of GT patients with moderate to severe bleeding treated with either rFVIIa alone or in combination with antifibrinolytic therapy and platelet transfusion, which effectively maintained the hemostasis and provided safety profile for most patients with a 72-91% success rate in stopping the bleeding [6]. Most of our patients responded well to rFVIIa combined with platelet transfusion.
Conclusions
Although GT rarely occurs worldwide but is common in Middle Eastern countries, GIAD or telangiectasia is not a common presentation of GT; however, some cases have been reported and require prompt treatment.
Acknowledgments
We would like to express our sincere gratitude to the Madinah Hereditary Blood Disorders Center for providing us with patient information to structure this article.
Financial Disclosure
The authors received no financial support for the research, authorship, and/or publication of this article.
Conflict of Interest
None to declare.
Informed Consent
Informed Consent was not applicable as data were provided directly from the registry of the Madinah Hereditary Blood Disorders Center.
Author Contributions
Raghad A. Tarawah wrote the full manuscript. Ahmad M. Tarawah, as the main responsible physician for all patients, provided the patients data from the Madinah Hereditary Blood Disorders Center Rejestry.
Data Availability
The data supporting the findings of this study are available from the corresponding author upon reasonable request.
| References | ▴Top |
- Nurden AT. Glanzmann thrombasthenia.
Orphanet J Rare Dis. 2006;1:10.
doi pubmed - Tarawah A, Owaidah T, Al-Mulla N, Khanani M, Elhazmi J,
Albagshi M, et al. Management of glanzmann’s Thrombasthenia - Guidelines based on an
expert panel consensus from Gulf Cooperation Council countries. Journal of Applied Hematology.
2019;10(1):1.
doi - Tarawah AM, Owaidah TM, Tarawah RA, et al. High prevalence
of a rare bleeding disorder: a report from glanzmann thrombasthenia registry of Al-Madinah,
Saudi Arabia. Blood. 2023;142(Supplement 1):396.
doi - Ai-Barghouthi SK, Ai-Othman A, Lardhi A. Glanzmann's
thrombasthenia-spectrum of clinical presentation on Saudi patients in the eastern province.
J Family Community Med. 1997;4(1):57-61.
pubmed - Di Minno G, Zotz RB, d'Oiron R, Bindslev N, Di Minno MN,
Poon MC, Glanzmann Thrombasthenia Registry I. The international, prospective Glanzmann
Thrombasthenia Registry: treatment modalities and outcomes of non-surgical bleeding episodes in
patients with Glanzmann thrombasthenia. Haematologica. 2015;100(8):1031-1037.
doi pubmed - Duarte BKL, de Souza SM, Costa-Lima C, Medina SS, Ozelo MC.
Thalidomide for the treatment of gastrointestinal bleeding due to angiodysplasia in a patient
with Glanzmann's thrombasthenia. Hematol Rep. 2017;9(2):6961.
doi pubmed - Marlu R, Barthelon J, Durand A, Mathieu N, Barro C, Granger
V, Tatu A, et al. Long-term therapy with bevacizumab in a patient with Glanzmann's
thrombasthenia and recurrent digestive bleeding due to gastrointestinal angiodysplastic lesions.
Am J Gastroenterol. 2015;110(2):352-353.
doi pubmed - Paciullo F, Fierro T, Calcinaro F, Zucca Giucca G, Gresele
P, Bury L. Long-term treatment with thalidomide for severe recurrent hemorrhage from intestinal
angiodysplasia in Glanzmann Thrombasthenia. Platelets. 2021;32(2):288-291.
doi pubmed - Barre A, Dreanic J, Flaujac C, Roussel-Robert V, Stieltjes N,
Combe-Marzelle S, Coriat R, et al. Is there a role for antiangiogenic therapy, bevacizumab, in
the treatment of recurrent digestive bleeding due to angiodysplasia in Glanzmann's
thrombasthenia? Haemophilia. 2016;22(4):e347-348.
doi pubmed - Coppola A, De Stefano V, Tufano A, Nardone G, Amoriello A,
Cerbone AM, Di Minno G. Long-lasting intestinal bleeding in an old patient with multiple mucosal
vascular abnormalities and Glanzmann's thrombasthenia: 3-year pharmacological management.
J Intern Med. 2002;252(3):271-275.
doi pubmed - Leach M, Makris M, Hampton KK, Preston FE. Norethisterone
therapy for bleeding due to gastrointestinal telangiectases in Glanzmann's thrombasthenia.
Br J Haematol. 1998;100(3):594-596.
doi pubmed - Nardone G, Rocco A, Balzano T, Budillon G. The efficacy of
octreotide therapy in chronic bleeding due to vascular abnormalities of the gastrointestinal
tract. Aliment Pharmacol Ther. 1999;13(11):1429-1436.
doi pubmed - Khosravi A, Rahimi H, Mansouritorghabeh H. Coincidence of
Glanzmann's thrombasthenia with hereditary haemorrhagic telengiectasia in a man with
gastrointestinal bleeding. Blood Coagul Fibrinolysis. 2015;26(1):98-100.
doi pubmed - Jackson CS, Strong R. Gastrointestinal Angiodysplasia:
Diagnosis and Management. Gastrointest Endosc Clin N Am. 2017;27(1):51-62.
doi pubmed
This
article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0
International License, which permits unrestricted non-commercial use, distribution, and
reproduction in any medium, provided the original work is properly cited.
Journal
of Medical Cases is published by Elmer Press Inc.